Pd-mediated carbene polymerisation: activity of palladium(II) versus low-valent palladium†
Nicole M. G.
Franssen
ab,
Joost N. H.
Reek
a and
Bas
de Bruin
*a
aVan't Hoff Institute for Molecular Sciences (HIMS), Department of Homogeneous and Supramolecular Catalysis, Universiteit van Amsterdam, P.O. box 94720, 1090 GS, Amsterdam, The Netherlands. E-mail: B.deBruin@uva.nl; Fax: +31 20 525 5604; Tel: +31 20 525 6495
bDutch Polymer Institute DPI, P.O. box 902, 5600 AX, Eindhoven, The Netherlands
First published on 12th November 2010
Abstract
Copolymerisation of olefins and carbene monomers was attempted with several well-defined Pd catalysts active in both olefin polymerisation and carbene polymerisation. In none of the attempts copolymer formation or even formation of the homopolymers was observed. This indicates that olefin polymerisation and carbene polymerisation are incompatible, despite the fact that the proposed transition states for these processes are very similar. Detailed investigations of Pd catalysed homopolymerisation of carbenes using both PdII and Pd0 complexes revealed that the active species in these reactions are most likely low-valent Pd species rather than PdII–alkyl species generally assumed to mediate carbene polymerisation. Well-defined PdII–alkyl species showed only a few insertions of carbene monomers, while longer oligomers (∼20 carbene units) are formed from Pd0 salts. In agreement with previous investigations, Pd0–NHC complexes allow formation of higher-Mw materials. Activation of the catalyst by NaBPh4 is required. Mechanistic studies revealed that involvement of PdII species in this process is highly unlikely, but the exact nature of the low-valent active species (Pd nanoparticles, molecular Pd0 or PdI species) is not clear. However, involvement of free radical species can be ruled out. Since olefin polymerisation requires PdII as the active species, the likely involvement of lower-valent Pd species in carbene polymerisation explains the incompatibility of both processes. The absence of formation of olefin homopolymers by well-known Pd-based olefin polymerisation catalysts in the presence of EDA can be explained by in situ reduction of the PdII species by EDA.
Introduction
Polymers bearing polar functionalities are an important class within the vast field of polymer chemistry, since they exhibit beneficial properties with respect to adhesion, paint/printability and surface properties.1 Commercial synthesis of these materials is mainly based on radical processes,2 which (so far) allow only a quite poor control over the stereo properties of the resulting polymers.3 Combined with a general lack of (relative) activity in the polymerisation of ethene and other olefins, this limits the applicability of controlled radical polymerisation techniques for the synthesis of functional (blocky co-)polymers with a defined microstructure and defined tacticities of the functional blocks.4 Such polymers are expected to reveal desirable material properties resulting from phase separation at the microscopic level.5A promising route towards such polymers is the use of lanthanide metallocene complexes and group 4 early transition metal metallocenes, and the like. These systems polymerise polar vinyl monomers via living coordination–addition polymerisation (metal-controlled anionic-polymerisation), allowing the controlled synthesis of both syndiotactic and isotactic (rich) polymers from methylacrylates, acrylates, (meth)acrylamides, acrylonitriles and vinyl ketones.6–8 Random copolymerisation of polar vinyl monomers and olefins with these methods still represents an unmet challenge, as is the catalytic synthesis (instead of synthesis via ‘stoichiometric’ living polymerisation) of stereochemically controlled polar vinyl polymers, which would allow a more economical production of stereoregular polymers from polar monomers.6
Substantial advances in the development of new late transition metal (LTM) catalysts for coordination–insertion-type polymerisation reactions have made it possible to prepare (random) copolymers of polar vinyl monomers and (aliphatic) alkenes, hence producing interesting new materials with varying microstructures (e.g. side-chain functionalised, main-chain functionalised, random and block copolymers).1,6,9 These catalytic systems produce more than one chain per metal-ion (chain transfer), and the most recently developed catalysts even allow a decent control over the amount of the incorporated functionalities. The field of transition metal catalyst development for both controlled homo- and copolymerisations has recently been reviewed by Takeuchi.10 Pd(diimine) complexes catalyse the copolymerisation of ethene with polar monomers resulting in highly branched structures.11 Linear copolymers could be obtained by applying (phosphino–sulfonato)Pd complexes as catalyst12 and recently it was shown that these complexes also show activity towards the homopolymerisation of polar monomers, although the number of sequential insertions was limited.13
Although the most recently developed catalysts allow a decent control over the amount of incorporated functionalities in the copolymerisation of polar/nonpolar monomers, the incorporation of the polar monomers is generally rather low.
Another challenge in this field is to use LTM catalysts in stereospecific polymerisation reactions. So far there are no reported examples of LTM catalysts capable of stereospecific polymerisation of polar vinyl monomers or stereospecific copolymerisation of polar vinyl monomers with olefins.6 Hence, despite all intriguing new developments there are still many important challenges to face in the development of efficient catalysts for the preparation of high molecular weight, (highly) stereoregular polymers with a tunable amount of polar functionalities.1,6,9
In this respect, C1 polymerisation (or ‘carbene’ polymerisation) techniques may offer valuable alternative synthetic methods for the synthesis of new materials that are not so easily accessible by the more common polymerisation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds,14–16 especially for the synthesis of densely functionalised carbon-chain polymers. We recently contributed to this field by showing that Rh-mediated carbene polymerisation, using diazoesters (ROC(O)CHN2) as the carbene precursors, allows the preparation of fully functionalised, high molecular weight, and highly syndiotactic carbon-chain polymers,17–20 in good yields.20 The resulting highly syndiotactic polymeric materials bear a polar functionality on every single carbon atom of the main chain.
C double bonds,14–16 especially for the synthesis of densely functionalised carbon-chain polymers. We recently contributed to this field by showing that Rh-mediated carbene polymerisation, using diazoesters (ROC(O)CHN2) as the carbene precursors, allows the preparation of fully functionalised, high molecular weight, and highly syndiotactic carbon-chain polymers,17–20 in good yields.20 The resulting highly syndiotactic polymeric materials bear a polar functionality on every single carbon atom of the main chain.
Due to similarities in their propagation mechanisms, combining carbene polymerisation with traditional olefin polymerisation could well be possible, perhaps allowing the synthesis of polar/nonpolar copolymers. Rh mediated carbene polymerisation proceeds via a cis-migratory carbene insertion mechanism into the Rh–C bond of the growing polymer chain, and the polymer tacticity is likely achieved by chain-end control.18 In essence, this mechanism is very similar to the mechanism of LTM catalysed alkene polymerisation/oligomerisation, as well as LTM catalysed olefin–CO copolymerisation reactions (Scheme 1). Hence, combination of both processes could be a viable way to obtain functionalised polymers. However, rather few Rh complexes show activity in olefin polymerisation and therefore the use of Rh as catalyst in olefin polymerisations is limited.21
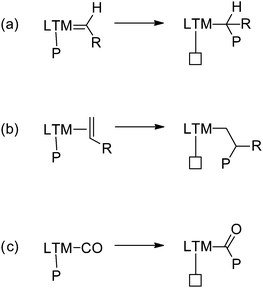 | ||
| Scheme 1 The mechanism of carbene polymerisation (a) shows similarities with that of olefin polymerisation (b) and olefin/CO copolymerisation (c). | ||
Pd seems the metal of choice for exploring the possibilities of carbene–olefin copolymerisation reactions, due to its known activity in both olefin and carbene homopolymerisation processes. Pd catalysts are amongst the most active LTM catalysts for (co)polymerisation reactions of both polar and non-polar vinyl monomers. The behavior of Pd in vinyl polymerisations has been studied thoroughly, both experimentally and computationally, and as a result the mechanism is well-understood (for a review see ref. 9b). Importantly, Pd catalysts are also active in the polymerisation of polar functionalised carbenes. PdII compounds active in carbene oligomerisation typically yield atactic and quite low-Mw materials,15 but some interesting new N-heterocyclic carbene (NHC) based Pd systems were recently developed that allow the formation of higher-Mw polymers. The applied NHC ligands were even reported to affect the polymer tacticities to some extent, thus showing possibilities for catalyst development by ligand design.22
In this paper we report our findings in the field of olefin–carbene copolymerisation reactions. We explored the reactivity of several well-defined Pd complexes towards ethyl diazoacetate (EDA) and ethene (Fig. 1).
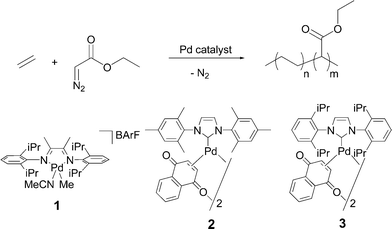 | ||
| Fig. 1 Top: hypothetical copolymerisation of ethene and ethyl diazoacetate (EDA). Bottom: structure of some Pd complexes (1–3) used in the attempted copolymerisation reactions. | ||
The results presented in this paper show that none of these Pd systems allow simultaneous alkene and carbene polymerisation. Hence carbene and alkene polymerisation reactions are incompatible (i.e. cannot be performed simultaneously or in the presence of the other monomer), despite the apparent similarities in their (proposed) reaction mechanisms (Scheme 1). We investigated some possible explanations for this behavior, which shed new light on Pd catalysed carbene (C1) homopolymerisation. A detailed investigation of these reactions is presented.
Results and discussion
We chose to start our investigations using the catalysts depicted in Fig. 1. Complex 1 is a PdII–diimine complex that is among the best LTM catalysts for ethene polymerisations, while Pd0 complexes 2 and 3 bearing N-heterocyclic carbene ligands were chosen as catalyst because of their reported high activity in EDA polymerisation allowing the formation of longer polycarbenes (Fig. 1).1. Olefin–carbene copolymerisation attempts
Attempts to copolymerise ethene and EDA in toluene using complex 1 failed, and no reaction occurred at all. Using higher ethene pressures (up to 10 bar) did not change this. No ethene or EDA homopolymers, neither any copolymers were formed under these conditions, and only unreacted EDA starting material could be recovered from the reaction mixture. This is quite surprising, because complex 1 is quite active in ethene homopolymerisation in the absence of EDA, and allows the copolymerisation of ethene with methylacrylate, in which case chelating carbonyl coordination is slowing down, but not blocking the polymerisation activity entirely. Apparently EDA does block ethene polymerisation completely, as under the applied copolymerisation conditions no polymer is formed at all. We performed some control experiments to confirm these observations, and thus we tested the use of complex 1 in the homopolymerisation of both ethene and EDA. In agreement with the abundant literature data for this system, also in our hands complex 1 proved a very active ethene polymerisation catalyst, thus reflecting its ‘normal behavior’ with respect to ethene. When the reaction was performed at an ethene pressure of 1 bar, polymers with a Mw up to 90 kDa were obtained after 3 h of reaction in either toluene or dichloromethane, emphasizing the high activity of this system towards ethene.
In contrast to our expectations, complex 1 does not homopolymerise EDA. GPC analysis indicated formation of only small amounts of low-Mw material (Mw < 900 Da, yield = 29%), which was not observed in the ethene–EDA copolymerisation attempt. Both ESI and MALDI-ToF mass analysis of this ‘oligomeric mixture’ showed peaks corresponding to palladium species with up to three [–C(COOEt)–] repeating units, suggesting the presence of the generalised structures shown in Fig. 2 (e.g. m/z = 707, n = 1).
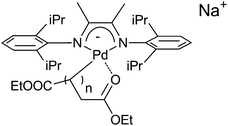 | ||
| Fig. 2 Generalised structure of the Pd species observed with ESI-MS (0 < n < 2). | ||
These species are likely formed upon insertion of EDA into a Pd–H bond, which in turn is likely formed from the starting Pd–Me complex through β-hydrogen elimination following single or multiple carbene insertion steps into the Pd–C bond.
Despite the cationic nature of the PdII-starting compound, the ESI mass data clearly reveal the presence of neutral [Pd(alkyl)(C(COOEt)n(diimine))] species (n = 1, 2 or 3), detected with Na+ as a charge carrier in the ESI-MS. This may suggest a reduction of the cationic [PdII(alkyl)(diimine)]+ material to neutral [Pd(alkyl)(diimine)] under the applied conditions. Although this implies a formal reduction of PdII to PdI, we prefer the ligand radical interpretation [PdII(alkyl)(diiminate˙−)] in view of the known redox non-innocence of diimine ligands.24–26 The reducing power of diazo compounds has been reported previously.28 Hence, it seems that reduction by EDA converts the catalytically active cationic Pd species into a non-active (neutral) ligand radical species, thus blocking both ethene and EDA polymerisation activity entirely. The combination of the reduced charge (lowering the probability of associative ligand substitution by the substrate) and coordination of the carbonyl groups of the few inserted carbene moieties to palladium may hamper the insertion of both ethene and carbene units.
Since ethene polymerisation and EDA polymerisation proved incompatible, we decided to investigate the possibility of EDA–styrene copolymerisation reactions. Since complex 1 shows no activity towards styrene due to formation of stable intermediates as a result of π-stacking, PdII–bipy complexes were applied in view of their reported activity in CO/styrene copolymerisation reactions. However, again no polymer was formed at all under the standard conditions generally applied for CO/styrene copolymerisation with this catalyst. Neither one of the homopolymers/oligomers was formed nor any sign of copolymer/oligomer formation was observed upon reaction of styrene with EDA in the presence of [(bipy)2Pd](PF6)2 in methanol as a solvent. Instead, only a small amount of EDA was consumed and several side products were formed (Fig. 3 and Table 1), most of which are indicative of carbene-transfer activity. Addition of a small amount of DMSO to keep the catalyst in solution led to a decreased activity, indicating that coordination of DMSO to the Pd center blocks reactions with EDA (entry 2). The formation of cyclopropanation product A and methanol-insertion product C is noteworthy. Both products indicate carbene-transfer activity and thereby in situ reduction of the PdII species to Pd0, since (mononuclear) Pd0 species are meanwhile known to be the most likely active species in Pd-mediated cyclopropanation reactions.27,28 The formation of the Δ2-pyrazoline product B formed by a [2 + 3] cycloaddition reaction is also catalysed, but this could well be the result of the organo-catalytic activity of liberated bipyridine rather than Pd,29 because formation of B was also observed when the reaction was carried out using a catalytic amount of pyridine in the absence of Pd. Changing the solvent from MeOH to DCM or MeCN did not lead to the formation of polymeric material either, although obviously this did prevent the formation of the methanol O–H insertion product. This product is formed upon insertion of EDA into the O–H bond of MeOH in a Pd catalysed reaction, and is observed as the only product when the reaction is performed in the absence of styrene.
| Entry | Catalyst | Solvent | Styrene (eq.) | EDA (eq.) | Products | Ratio A(cis)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :A(trans):Bb :A(trans):Bb |
|---|---|---|---|---|---|---|
| a Reaction conditions: solvent (1 mL), styrene, EDA, and Pd (9 µmol), 16 h reaction time. b Product C was removed in vacuo. c A mixture of catalyst and styrene in MeOH was treated with CO for 1 min before addition of EDA; CO flow was stopped after 8 min while the reaction was stirred for an additional 16 h. | ||||||
| 1 | [(bipy)2Pd](PF6)2 | MeOH | 2000 | 2000 | A, B, C | 0.60:0.45:1 |
| 2 | [(bipy)2Pd](PF6)2 | MeOH/DMSO (50:1) |
2000 | 2000 | A, B, C | 0.16:0.32:1 |
| 3 | [(bipy)2Pd](PF6)2 | DCM | 2000 | 2000 | A, B | 0.55:0.58:1 |
| 4 | [(bipy)2Pd](PF6)2 | MeCN | 2000 | 2000 | A, B | 7:4:1 |
| 5 | [(bipy)Pd(Me)(MeCN)]PF6 | MeOH | 2000 | 2000 | C | — |
| 6 | [(bipy)Pd(Me)(MeCN)]PF6 | — | 2000 | 2000 | No reaction | — |
| 7c | [(bipy)Pd(Me)(MeCN)]PF6 | MeOH | 60000 |
60000 |
No reaction | — |
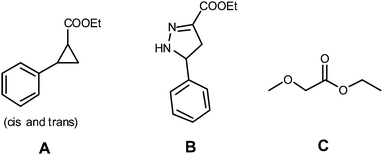 | ||
| Fig. 3 Observed products formed in the attempted styrene/EDA copolymerisation mediated by Pd(bipy)L2. | ||
[(bipy)Pd(Me)(MeCN)]PF6 is a pre-activated form of the above [(bipy)2Pd]2+ catalyst, but this compound shows no (co)polymerisation activity in these reactions either. Hence, the lack of reactivity does not stem from any poor activation/initiation efficiency. Reaction of EDA and styrene with [(bipy)Pd(Me)(MeCN)]PF6 in MeOH led to formation of only product C. When the reaction was carried out in the absence of solvent no reaction occurred at all. The limited role of catalyst (pre)activation problems is further emphasised by the results obtained in entry 7, in which the catalyst is pre-treated with CO in the presence of styrene in order to form a Pd–acyl species (a known intermediate in CO/styrene copolymerisation), which in the presence of EDA again does not produce any polymer at all.
The above observations, together with the above described lack of reactivity of well-defined PdII–alkyl complexes towards EDA (co)polymerisation (Section 1.1), are rather surprising, because PdII–alkyl species were proposed to be the key active species in carbene polymerization reactions from substituted diazo compounds.14,15,22 Taken together, the results suggest that lower oxidation state Pd species may well play an important role in carbene polymerisation mediated by Pd. This prompted us to re-investigate the reported EDA homopolymerisation reactions mediated by Pd species, in which we focused on the possible role of low-valent species in these reactions.
2. Palladium mediated homopolymerisation of EDA revisited
| Entry | Catalyst | Solvent | T/°C | M n b/Da | M n/Mwb | Yield (%) |
|---|---|---|---|---|---|---|
| a Reaction conditions: solvent (5 mL), EDA (0.95 mmol, 100 eq.), and Pd (9 µmol); 16 h reaction time. b Determined by GPC in DCM at 35 °C vs. polystyrene. c Isolation of the product hampered by work-up. d 50 eq. of EDA were used. | ||||||
| 1 | 3 | DCM | RT | 900 | 1.06 | 42 |
| 2 | 4 + MAO (100 eq.) | Toluene | RT | 1100 | 1.22 | n.d.c |
| 3 | 5 | DCM | RT | 1100 | 1.21 | 61 |
| 4 | (cod)PdMeCl + NaBArF | DCM | RT | 1500 | 1.32 | 29 |
| 5 | (cod)PdMeCl | DCM | RT | 2000 | 1.28 | 53 |
| 6 | Pd(dba)2 | DCM | 40 | 1600 | 1.34 | 72 |
| 7 | Pd(dba)2 | CHCl3 | 40 | 1400 | 1.50 | 59 |
| 8 | Pd(dba)2 | Toluene | 40 | 1400 | 1.21 | 62 |
| 9d | Pd(dba)2 | Toluene | 40 | 1700 | 1.33 | 71 |
| 10 | Pd(dba)2 | Toluene | RT | 1300 | 1.18 | 52 |
| 11 | Pd(dba)2 + 1 eq. NaBPh4 | Toluene | 40 | 1500 | 1.27 | 74 |
| 12 | Pd2(dba)3 | DCM | 40 | 2000 | 1.20 | 80 |
| 13 | (nbd)Pd(ma) | DCM | 40 | 1500 | 1.22 | 78 |
| 14 | Pd/C | DCM | 40 | 1100 | 1.08 | 74 |
PdII–diimine complexes 1, 4 and 5 (Fig. 1 and 4) showed only few insertions of EDA, giving rise to low-Mw material that consisted mainly of Pd complexes with structures similar to that in Fig. 2 (Section 1.1). Not nearly all EDA is converted and the overall yield of these reactions is rather low. In situ activation of the catalyst by methylaluminoxane (MAO) in toluene did not lead to a significant increase of the Mw and again the major components in the mixture were Pd complexes obtained upon few EDA insertions (entry 2 vs. 1). Using THF as solvent for the reaction of complex 4 and MAO led to the formation of substantial amounts of polymeric material, which could, however, be identified as poly-THF.30 Aside from this, no oligomers or polymers from EDA could be detected. Complex 5 gave rise to a product mixture similar to that of complex 1, indicating that the results were not affected by changing the steric bulk around the metal center (Table 2, entry 3 vs. 1). This indicated that the ligand does not play an important role during the catalysis and therefore (1,5-cyclooctadiene)PdII complexes were tested as catalyst as well. In situ activation of (cod)PdMeCl by NaBArF did lead to a minor increase in Mw, but the yield dropped significantly (Table 2, entry 4). Surprisingly, this catalyst performed better, both in terms of yield and Mw of the oligomers when it was applied without activation by NaBArF (NaCl precipitation), despite the fact that this catalyst does not have a readily available vacant site (Table 2, entry 5).
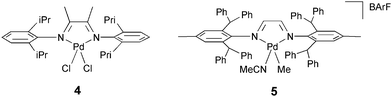 | ||
| Fig. 4 PdII–diimine complexes 4 and 5 applied as catalyst in EDA homopolymerisation. | ||
All data gathered so far suggest the involvement of in situ generated low-valent Pd species in EDA polymerisation. All studied Pd0 sources performed better in terms of yield (Table 2, entries 5 to 14) and in these cases the main part of the reaction mixture consisted of oligomers detached from the Pd centre, with values for Mw listed in Table 2. Changing the reaction conditions led to minor improvements in yield and Mw. Pd2(dba)3 turned out to be a quite decent catalyst for this reaction in terms of oligomer yield; however, still only atactic and very low Mw material was obtained. In line with the suggested activation of Pd0 sources by BPh4 salts to yield higher-Mw polymers as reported by Ihara,22 the polymerisation of EDA with Pd(dba)2 in the presence of NaBPh4 was attempted as well. For this catalyst, however, BPh4 has no influence at all.
The likely involvement of low-valent Pd species as the active species in EDA polymerisation was further underlined by a comparable activity of heterogeneous metallic Pd dispersed on carbon (entry 14). The filtrate that remained after removal of the catalyst by filtration over Celite showed no residual catalytic activity after addition of additional fresh EDA, suggesting that the oligomers in this case are formed by the heterogeneous Pd0 material on carbon, and not likely by homogeneous (molecular) Pd species leached into solution. The heterogenic nature of the active species was further confirmed by performing Crabtree's test using dibenzo(a,e)cyclooctatetraene (dct) to inhibit possible homogeneous species.31 In the presence of 4 equivalents of dct (w.r.t. Pd) oligomers were formed with yields and Mw comparable to the reaction in the absence of dct, indicating that heterogeneous Pd0 species are responsible for the catalysis. Upon recycling the heterogeneous catalyst the oligomer yield drops to 10% after the second run and the activity is almost negligible after the third run. In all runs the molecular weight distribution of the product is similar.
To rule out the participation of heterogeneous Pd particles in the reactions catalysed by originally homogenous Pd0 complexes we studied the activity of Pd(dba)2 in the presence of mercury. Larger heterogeneous Pd species are known to be inactive in the presence of mercury. Since this test is not always conclusive in the case of catalysis by smaller (homogeneously dispersed) palladium nanoparticles, Crabtree's test was performed as well as a complementary test to inhibit possible homogeneous species.31 These experiments clearly showed that contribution of larger heterogeneous Pd species to the catalysis is highly unlikely, since catalyst performance in both terms of yield and Mw was similar in the presence or in the absence of mercury. When the catalyst was allowed to interact with dct for 2 hours before addition of EDA, the initially deep red mixture turned black, probably due to the formation of heterogeneous Pd black. This black mixture showed no reactivity towards EDA, indicating that Pd black is not the active species in these reactions either.
The formation of Pd black is clearly triggered by the presence of dct and does not visibly occur in the reactions without dct. The experiments do not rule out the participation of homogeneously dispersed Pd nanoparticles. The involvement of molecular homogeneous Pd0 species in the EDA polymerisation, however, seems to be the most probable explanation for these results.
The 1H NMR spectra of the oligomers showed similar patterns regardless of the catalyst used, indicating that the ligand environment around the catalyst has no significant influence on the oligomer microstructure. In all cases very broad resonances were observed, indicative for atactic material. This is in agreement with the spectra of oligomers obtained by PdII salts as catalyst reported previously by Ihara and coworkers.15 Resonances corresponding to oligomer end groups were not observed in either 1H or in 13C NMR.
MALDI-ToF mass analysis in reflector mode of the product mixtures obtained with either (cod)PdCl2 as a PdII source or Pd(dba)2 or (nbd)Pd(ma) as Pd0-sources showed clearly the presence of the oligomeric products detached from Pd (Fig. 5). This is in contrast to the observation of Pd-containing oligomeric species formed in the presence of PdII(diimine) catalysts (vide supra). All spectra revealed that the structures are built from [–C(COOEt)–] repeating units (Δm/z = 86) regardless of the exact catalyst used. Aside from this, [–C(COOH)–] units were observed when the reaction mixtures were subjected to acidified MeOH in order to precipitate Pd black. Fragments containing the same number of repeating units gave rise to various series of m/z signals, all with the general structure [Y–(C(COOEt))n–(C(COOH))m–Z]. The most dominant peaks were observed for Y = –C(COOEt)C(COOEt)H, Z = –OH and Y = –C(COOEt)C(COOEt)H, Z = –H. This indicates that the main chain-termination or chain-transfer pathway proceeds via β-hydrogen elimination (Scheme 2). Interestingly, when (nbd)Pd(ma) was used as catalyst, the incorporation of the maleic anhydride in the oligomer chain was observed (Y = HOC(O)CCC(O)O–, Z = –H and Y = HOC(O)CCC(O)O–, Z = –OH).
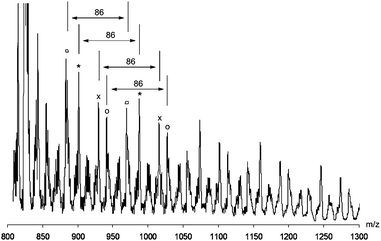 | ||
| Fig. 5 MALDI-ToF spectrum of oligomers obtained in the reaction of EDA with Pd(dba)2. | ||
 | ||
| Scheme 2 Schematic representation of chain transfer via β–hydrogen elimination. | ||
The above data clearly suggest the involvement of low-valent Pd species as the active species in carbene homopolymerisation from EDA. This also readily explains the incompatibility of carbene polymerisation with olefin polymerisation, as the latter is known to be catalysed by PdII–alkyl species. Since the Pd0 and PdII sources produce the same type of low Mw and atactic oligomers with comparable PDI values, combined with the above indications for reduction of PdII to lower-valent species by EDA, we herewith postulate that the observed activity of the PdII sources towards EDA homopolymerisation is most likely due to in situ reduction of PdII by EDA.
In agreement with the experiments described by Ihara, EDA is indeed converted to higher mass PEA polymer by the catalytically active species generated by mixing complex 3 with NaBPh4 in THF. Atactic PEA with a Mw up to 34 kDa can be obtained with this system (Table 3, entry 1). However, in addition to the polymer, also low-Mw oligomeric species (Mw < 1500 Da) are formed in this reaction, thus indicating that the catalyst system contains more than one active species. Without access to preparative-recycling GPC (PR-GPC), as applied by Ihara, these oligomers cannot easily be separated from the polymers. In Table 3 we therefore report the combined yields of these polymeric and oligomeric products. The reported Mn and Mw/Mn values listed correspond to the polymeric fraction. Using a catalyst loading of ∼1 mol% with respect to EDA leads to a combined yield of 80% (entry 1). If we compare this value with the much lower reported PEA polymer yield obtained under similar reaction conditions by Ihara after isolation by PR-GPC (50%), it becomes clear that the oligomers are present in significant amounts (∼30%).
| Entry | Catalyst | Cocatalyst | M n b/Da | M w/Mnb | Yieldc (%) |
|---|---|---|---|---|---|
| a Reaction conditions: THF (2 mL), catalyst (4.5 µmol), cocatalyst (11 µmol) and EDA (0.95 mmol). b M w determined by GPC in DCM at 35 °C vs. polystyrene. c Combined yield of oligomers and polymers. | |||||
| 1 | 3 | NaBPh4 | 34000 |
1.38 | 80 |
| 2 | 2 | NaBPh4 | 23000 |
1.92 | 95 |
| 3 | 3 | Bu4NPF6 | 3700 | 1.07 | 58 |
| 4 | 3 | — | 3000 | 1.38 | 86 |
| 5 | 3 | BPh3 | 2960 | 1.02 | 49 |
| 6 | — | BPh3 | 800 | 1.11 | <5 |
In good agreement with the reported data, changing the substituents on the NHC ligand of the catalyst from 2,6-diisopropylphenyl to mesityl (entry 2) leads to somewhat shorter polymers in a slightly increased combined yield. The influence of the NHC ligand substituents on the outcome of the polymerisation reaction (Table 3, entries 1 and 2) suggests that the NHC ligand remains coordinated to the active species responsible for the polymer formation. However, in contrast with the reported data this catalyst gives rise to a broader molecular weight distribution in our hands. A further contrasting observation with the reported data is that we could not reproduce the previously observed influence of the NHC ligand on the polymer tacticity. 1H and 13C NMR analysis of the crude product mixtures obtained with the two different ligands revealed exactly the same spectra, each indicative of atactic material. At this point we cannot rule-out that these differences can be explained by the fact that we did not separate the PEA polymer fraction from the oligomeric side products (in contrast to what was reported), but we would at least have expected to see the reported sharp signals of the syndiotactic enrichments in the NMR spectra.
Actually, the NMR data of the oligomers and polymers produced by all of the different Pd-catalysts (re)investigated in this report are remarkably similar to those of atactic PEA prepared by radical polymerisation of diethylfumarate. This raises the question to what extent Pd-initiated radical polymerisation plays a role in these reactions (vide infra).
When the reactions were performed in the absence of tetraaryl borate salts, only low Mw oligomers were formed, indicating that the borate is indeed involved in generating the active species. Other salts, such as [Bu4N]PF6, are not effective in promoting the formation of higher Mw polymeric material, and hence this is not a general salt effect (Table 3, entry 3). The GPC trace of this reaction showed the presence of multiple low-Mw species, of which most have a Mw < 1500 Da. Similar results are obtained when the reaction is carried out without any cocatalyst (Table 3, entry 4).
1H NMR analysis of the catalyst mixture in THF under inert conditions reveals within a few minutes the appearance of some small additional peaks indicating the formation of (a) different complex(es). From the spectra it is clear that there is some reaction going on between the Pd–NHC species and BPh4−, but whether or not this (these) species is (are) involved in the carbene polymerization is not clear. The NMR spectrum is clearly dominated by unreacted BPh4− and the Pd–NHC starting compound, and the additional peaks do not rise much in intensity after a few hours at RT under inert conditions. The NMR spectra are therefore not conclusive for the nature of the active species. Mass analysis of the catalyst mixture in the presence of NaBPh4 indicated that (some) phenyl group transfer from the borate to palladium occurs. The ESI-MS spectrum of the catalyst mixture showed peaks corresponding to [(NHC)Pd(phenyl)]H+ (m/z = 573), and (NHC + phenyl)+ (m/z = 465), suggesting that the phenyl group is being transferred via Pd to end up finally at the NHC ligand by reductive elimination. The observation of a peak corresponding to (NHC–H)+ (m/z = 389) indicates that the Pd easily looses the NHC ligand under these ESI-MS conditions. These results are confirmed by FAB measurements, suggesting that the observed phenyl transfer and the observation of free ligand are not induced by the conditions of the mass analysis. Control experiments using BPh3 instead of NaBPh4 as cocatalyst showed only formation of oligomers (Table 3, entry 5). Almost no activity was observed if the reaction was performed using only BPh3 in the absence of Pd species 3, and formation of oligomers was not observed (Table 3, entry 6). This confirms that the catalytically active species are Pd based, excluding the possible activity of in situ generated boranes (BPh3).14,16 It should be noted here that in analogy with phenyl transfer from phenylboronic acids, phenyl transfer from BPh4− likely proceeds via a direct phenyl-anion transfer to the metal. Such transmetallation processes generally do not require a change in the oxidation state of the transition metal.32 Hence, the reaction medium is more likely to be reductive rather than oxidative, and there is no good reason to assume the formation of PdII–alkyl species from the Pd0 starting material under these conditions. Therefore, we speculate that also in this case low-valent (NHC)Pd–phenyl species (Pd0 or perhaps PdI) are the more likely carbene polymerisation initiating and mediating species.
Radical trap experiments. Since the polymers obtained with the above NHC–Pd0 species reveal remarkably similar NMR spectra to those obtained by radical polymerisation of diethylfumarate, we decided to investigate the possibility of Pd triggered radical polymerisation processes.33 Phenyl transfer to palladium has in some cases been reported to trigger the formation of radical species,34 and the involvement of Pd radical species in several catalysed reactions is abundant. This topic has recently been highlighted by Ford and Jahn.35 Hence, we decided to perform the EDA polymerisation reaction in the presence of several different reagents known to function as radical traps (Table 4).
| Entry | Additive | [Add]/[Pd] | M n b/Da | M w/Mnb | Yieldc (%) |
|---|---|---|---|---|---|
| a Reaction conditions: THF (2 mL), 3 (4.5 µmol), NaBPh4 (11 µmol) and EDA (0.95 mmol, 100 eq.). b M w determined by GPC in DCM at 35 °C vs. polystyrene. c Combined yield of oligomers and polymers. d TEMPO = 2,2,6,6-tetramethylpiperidine-1-oxyl. e TBP = 2,6-di-tert-butylperoxide. f HQ = hydroquinone. | |||||
| 1 | None | — | 34000 |
1.38 | 80 |
| 2 | TEMPOd | 2 | 38000 |
1.45 | 56 |
| 3 | TEMPO | 40 | 37000 |
1.47 | 59 |
| 4 | TBPe | 0.7 | 44000 |
1.38 | 67 |
| 5 | HQf | 0.8 | 39000 |
1.64 | 84 |
| 6 | HQ | 20 | 36000 |
1.42 | 65 |
| 7 | MeCN | 40 | 60000 |
1.53 | 81 |
EDA polymerisation catalysed by complex 3 is in no case completely blocked by the presence of the several different radical traps listed in Table 4, although TEMPO does seem to have a small influence by decreasing the polymer yield somewhat. Higher amounts of TEMPO, however, do not further reduce the yields, and TEMPO does not seem to have significant influence on the polymer length or PDI (Table 4, entries 2 and 3 vs. entry 1).
All other radical traps tested led to polymers in good yields with equal or even slightly higher Mw compared to the reaction without the traps (Table 4, entry 1). Addition of small amounts of hydroquinone (HQ) led to a slightly higher Mw (Table 4, entry 5). Adding more equivalents of this trap did not lead to significant changes, although the polymer yield dropped slightly (Table 4, entries 5 and 6). The slight increase of Mw observed for some of the traps can be explained by coordination of the radical traps to a vacant site of Pd, thereby somehow stabilizing the resting state of the active catalyst. This is emphasised by the substantially increased Mw obtained if the reaction is performed in the presence of 40 eq. acetonitrile (Table 4, entry 7).
Although in some cases the polymer yield is affected by the radical traps, the results are not indicative of a radical polymerisation process, which we would expect to be completely blocked by the applied radical traps.
Mercury and Crabtree's tests. Regarding the likely role of low-valent Pd species, and the fact that heterogeneous Pd/C is active in EDA oligomerisation, we next focused on the possibility that the applied NHC–Pd0 species might generate heterogeneous (colloidal) palladium (nano)particles being the real active species in these reactions. This would also readily explain the lack of reactivity of this system towards ethene.
To study this possibility in more detail, also the reaction with this catalyst was subjected to both the mercury test and the complementary Crabtree's test. Fig. 6 clearly shows that the contribution of larger heterogenous palladium particles to the catalysis is highly unlikely for this catalyst, since almost no effect of mercury on the polymer length and yield is observed.
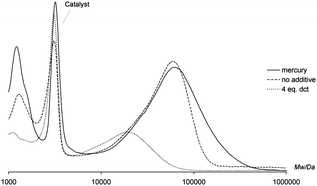 | ||
| Fig. 6 Effect of addition of mercury and dct on polymer formation. (a) Reaction conditions: THF (2 mL), 3 (4.5 µmol), NaBPh4 (11 µmol), EDA (0.95 mmol, 100 eq.), and additive (1 drop Hg or 4 eq. dct), 16 h reaction time; (b) Mw determined by GPC at 35 °C vs. polystyrene. | ||
This is underlined by the observation of a significant decrease in polymer length when the reaction is performed in the presence of dct, indicating that the catalytically active species is most likely homogeneous in nature.
Based on the combined above-mentioned results, we can conclude that the NHC–Pd0 species are behaving quite differently in EDA polymerisation compared to other Pd sources. Most Pd compounds convert EDA to low molecular weight atactic oligomers, all of the same length and with comparable PDI values, thus suggesting that they all generate the same or very similar active Pd species. Since heterogeneous Pd/C gives virtually the same results, ascribing the activity to low-valent Pd seems most likely for all these Pd sources. Molecular Pd0 species or Pd0 (nano)clusters are likely candidates, but PdI intermediates cannot be completely ruled out. The NHC–Pd0 precursors produce higher molecular weight material, and these reactions are clearly catalysed by (a) homogeneous catalyst(s). The apparent influence of the NHC ligand on the catalytic performance is interesting from a ligand-design perspective.
Conclusions
Synthesis of functionalised polymers by combining carbene polymerisation with olefin polymerisation in a Pd catalysed reaction is hampered by a difference in active species for both homopolymerisation reactions. Despite good activity of the catalysts used in homopolymerisation of either one of the monomers, formation of carbene–olefin copolymers never occurred (for none of the catalysts attempted). Remarkably, both homopolymerisation reactions are completely blocked in the presence of the other comonomer, showing that carbene and olefin polymerisations are fully incompatible.Detailed investigations of the homopolymerisation of EDA under several conditions and using several different Pd sources led us to conclude that the active Pd in this reaction is likely some low-valent Pd material, either homogenous molecular Pd, (colloidal) Pd metal (nano)clusters or even heterogenous Pd black. The Pd0–NHC/NaBPh4 systems recently reported by Ihara seem to get activated by phenyl group transfer from the borate to Pd. Mercury and Crabtree's tests showed that this is most likely a molecular Pd0 catalyst. The performance of these systems is clearly influenced by the nature of the NHC ligand, which is interesting from a ligand-design perspective, and could eventually lead to the development of Pd catalyst that allows the synthesis of higher mass and possibly stereoregular polycarbenes. However, so far all known Pd systems still only produce atactic materials, and unfortunately do not allow the copolymerisation of EDA with olefins, which was our main interest in these catalysts. Hence, also for these systems it seems quite unlikely that the polymerisation proceeds via discrete PdII–alkyl intermediates, for which we would have expected at least some olefin incorporation. This is emphasised by the higher activity of other Pd0 sources compared to PdII species in EDA oligomerisation. The lack of activity of well defined Brookhart-type PdII–alkyl species in EDA polymerisation further speaks against the involvement of PdII active species in these reactions. The exact nature of the Pd catalyst remains elusive, as is the case in many Pd catalysed reactions. Molecular Pd0 species or Pd0 (nano)clusters are likely candidates, but PdI intermediates cannot be completely ruled out. The polycarbenes are, however, not formed by a radical polymerisation process.
Experimental section
All manipulations were performed under nitrogen atmosphere using standard Schlenk conditions. Methanol and dichloromethane were distilled from CaH2. Toluene was distilled from sodium and THF from sodium/benzophenone. α-Diimine ligands (2,6-iPrC6H3)NC(Me)–C(Me)N(2,6-iPrC6H3),36 (2,6-bis(diphenylmethyl)-4-methylphenyl)–NC(Me)–C(Me)N(2,6-bis(diphenylmethyl)-4-methylphenyl),37 (2,6-iPrC6H3)NC(Me)–C(Me)N(2,6-iPrC6H3)PdCl238 and [(2,6-iPrC6H3)NC(Me)–C(Me)N(2,6-iPrC6H3)–Pd(Me)(MeCN)]BArF39 were prepared according to literature procedures. [(2,6-Bis(diphenylmethyl)-4-methylphenyl)NC(Me)–C(Me)N(2,6-bis(diphenylmethyl)-4-methylphenyl)Pd(Me)(MeCN)]BArF was prepared in a similar manner as Pd–bian systems.40 The synthesis of (cod)Pd(Me)(Cl),41 [(bipy)2Pd](PF6)2,42 and (nbd)Pd(ma)43 have been reported previously. [(bipy)Pd(Me)(MeCN)]PF6 was synthesised in analogy with the Pd–diimine complexes.41 Styrene was purified over basic alumina prior to use. All other chemicals were purchased from chemical suppliers and used as received without further purification. NMR analyses were carried out on Varian Mercury 300 spectrometer (300 MHz for 1H and 75 MHz for 13C, respectively). Molecular mass distributions were measured using size exclusion chromatography (SEC) on a Shimadzu LC-20AD system with two PLgel 5 µm MIXED-C columns (Polymer Laboratories) in series and a Shimadzu RID-10A refractive index detector, using dichloromethane as a mobile phase at 1 mL min−1 and T = 35 °C. Polystyrene standards in the range of 760–1880000 g mol−1 (Aldrich) were used for calibration. Molecular mass distributions for polyethene were measured using size exclusion chromatography (SEC) on a Shimadzu LC-10AD system with three PLgel 10 µm MIXED-B columns (Polymer Laboratories) in series, in combination with a Shimadzu SIL 9A autoinjector and a Dawn DSP-F MALS photometer, using THF as a mobile phase at 1 mL min−1 and T = 35 °C.
Ethene polymerisations
A solution of Pd catalyst (9 µmol) in toluene (10 mL) was pressurised with ethene (1 bar). After stirring for 3 h at room temperature the mixture was vented and the product was isolated by precipitation in MeOH. Mw = 90 kDa.General procedure for EDA–ethene copolymerisation reactions
A solution of Pd catalyst (9 µmol) in solvent (2 or 10 mL) was pressurised with ethene (1 bar) prior to addition of the desired amount of EDA. After stirring for 3 h at room temperature the mixture was vented. Removal of the liquids in vacuo yielded the crude products.General procedure for EDA–styrene copolymerisation reactions
Styrene (250 µL, mmol) was added to a solution of Pd catalyst in solvent (1 mL). After addition of EDA (200 µL, mmol) the mixture was stirred overnight at room temperature. Removal of the liquids in vacuo yielded the crude product mixture.General procedure for EDA homopolymerisation reactions
To a solution of Pd catalyst (9 µmol) in solvent (5 mL) was added EDA (100 µL, 0.95 mmol) and the mixture was stirred overnight. Removal of the volatiles in vacuo yielded the crude product mixture as yellow oil.Acknowledgements
We kindly thank Jeroen Wassenaar and Guillaume Berthon-Gelloz for providing their complexes and ligands, Petra Aarnoutse for carrying out the MALDI-ToF analysis, and Tom Aalbers for the analyses of our polyethene samples with GPC. This project is part of the research program of the Dutch Polymer Institute DPI, Eindhoven, The Netherlands (projects #646 and #647).References
- A. Nakamura, S. Ito and K. Nozaki, Chem. Rev., 2009, 109, 5215 CrossRef CAS.
- Encyclopedia of Polymer Science and Engineering, ed. H. F. Mark, N. M. Bikales, C. G. Overberger and G. Mendes, Wiley, New York, 1986, vol. 13, p. 708 Search PubMed.
- K. Satoh and M. Kamigaito, Chem. Rev., 2009, 109, 5120 CrossRef CAS.
- M. Ouchi, T. Terashima and M. Sawamoto, Chem. Rev., 2009, 109, 4963 CrossRef CAS.
- H.-C. Kim, S.-M. Park and W. D. Hinsberg, Chem. Rev., 2010, 110, 146 CrossRef CAS.
- E. Y.-X. Chen, Chem. Rev., 2009, 109, 5157 CrossRef CAS.
- (a) S. Collins and D. G. Ward, J. Am. Chem. Soc., 1992, 114, 5460 CrossRef CAS; (b) A. D. Bolig and E. Y.-X. Chen, J. Am. Chem. Soc., 2001, 123, 7943 CrossRef CAS; (c) H. Deng, T. Shiono and K. Soga, Macromolecules, 1995, 28, 3067 CrossRef CAS; (d) C. Cui, A. Shafir, C. L. Reeder and J. Arnold, Organometallics, 2003, 22, 3357 CrossRef CAS.
- W. E. Goode, F. H. Owens, R. P. Fellman, W. H. Snyder and J. E. Moore, J. Polym. Sci., 1960, 46, 317–331 CrossRef.
- For recent reviews on coordination–insertion copolymerisation of polar monomers, see: (a) L. S. Boffa and B. M. Novak, Chem. Rev., 2000, 100, 1479 CrossRef CAS; (b) S. D. Ittel, L. K. Johnson and M. Brookhart, Chem. Rev., 2000, 100, 1169 CrossRef CAS.
- D. Takeuchi, Dalton Trans., 2010, 39, 311 RSC.
- L. K. Johnson, S. Mecking and M. Brookhart, J. Am. Chem. Soc., 1996, 118, 267 CrossRef CAS.
- E. Drent, R. van Dijk, R. van Ginkel, P. van Oort and R. I. Pugh, Chem. Commun., 2002, 744 RSC.
- D. Guironnet, P. Roesle, T. Rünzi, I. Göttker-Schnetmann and S. Mecking, J. Am. Chem. Soc., 2009, 131, 422 CrossRef CAS.
- E. Jellema, A. L. Jongerius, J. N. H. Reek and B. de Bruin, Chem. Soc. Rev., 2010, 39, 1706 RSC.
- (a) E. Ihara, M. Fujioka, N. Haida, T. Itoh and K. Inoue, Macromolecules, 2005, 38, 2101–2108 CrossRef CAS; (b) E. Ihara, M. Kida, M. Fujioka, N. Haida, T. Itoh and K. Inoue, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 1536–1545 CrossRef CAS; (c) E. Ihara, A. Nakada, T. Itoh and K. Inoue, Macromolecules, 2006, 39, 6440–6444 CrossRef CAS; (d) E. Ihara, T. Hiraren, T. Itoh and K. Inoue, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 1638 CrossRef CAS; (e) E. Ihara, T. Hiraren, T. Itoh and K. Inoue, Polym. J. (Tokyo), 2008, 40, 1094 Search PubMed; (f) E. Ihara, N. Haida, M. Iio and K. Inoue, Macromolecules, 2003, 36, 36 CrossRef CAS; (g) E. Ihara, Y. Goto, T. Itoh and K. Inoue, Polym. J. (Tokyo), 2009, 41, 1117 Search PubMed.
- (a) K. J. Shea, J. W. Walker, H. Zhu, M. M. Paz and J. Greaves, J. Am. Chem. Soc., 1997, 119, 9049 CrossRef CAS; (b) B. B. Busch, M. M. Paz, K. J. Shea, C. L. Staiger, J. M. Stoddard, J. R. Walker, X.-Z. Zhou and H. Zhu, J. Am. Chem. Soc., 2002, 124, 3636 CrossRef; (c) K. J. Shea, B. B. Busch and M. M. Paz, Angew. Chem., Int. Ed., 1998, 38, 1391 CrossRef; (d) C. E. Wagner, J.-S. Kim and K. J. Shea, J. Am. Chem. Soc., 2003, 125, 12179 CrossRef CAS; (e) C. E. Wagner, A. A. Rodriguez and K. J. Shea, Macromolecules, 2005, 38, 7286 CrossRef CAS.
- D. G. H. Hetterscheid, C. Hendriksen, W. I. Dzik, J. M. M. Smits, E. R. H. van Eck, A. E. Rowan, V. Busico, M. Vacatello, V. Van Axel Castelli, A. Segre, E. Jellema, T. G. Bloemberg and B. de Bruin, J. Am. Chem. Soc., 2006, 128, 9746–9752 CrossRef CAS.
- E. Jellema, P. H. M. Budzelaar, J. N. H. Reek and B. de Bruin, J. Am. Chem. Soc., 2007, 129, 11631–11641 CrossRef CAS.
- M. Rubio, E. Jellema, M. A. Siegler, A. L. Spek, J. N. H. Reek and B. de Bruin, Dalton Trans., 2009, 8970 RSC.
- E. Jellema, A. L. Jongerius, A. J. C. Walters, J. M. M. Smits, J. N. H. Reek and B. de Bruin, Organometallics, 2010, 29, 2823–2826 CrossRef CAS.
- J. Sedláček and J. Vohlídal, Collect. Czech. Chem. Commun., 2003, 68, 1745 CrossRef CAS.
- E. Ihara, Y. Ishiguro, N. Yoshida, T. Hiraren, T. Itoh and K. Inoue, Macromolecules, 2009, 42, 8608 CrossRef CAS.
- P. Margl and T. Ziegler, J. Am. Chem. Soc., 1996, 118, 7337 CrossRef.
- L. Johansson, O. B. Ryan, C. Rømming and M. Tilset, Organometallics, 1998, 17, 3957 CrossRef CAS.
- A. C. Bowman, C. Milsmann, E. Bill, E. Lobkovsky, T. Weijhermüller, K. Wieghardt and P. J. Chirik, Inorg. Chem., 2010, 49, 6110 CrossRef CAS.
- B. de Bruin, D. G. H. Hetterscheid, A. J. J. Koekkoek and H. Grützmacher, Prog. Inorg. Chem., 2007, 55, 247.
- G. Berthon-Gelloz, M. Marchant, B. F. Straub and I. E. Markó, Chem.–Eur. J., 2009, 15, 2923 CrossRef CAS.
- O. Illa, C. Rodríguez-García, C. Acosta-Silva, I. Favier, A. Oliva, M. Gómez, V. Branchadell and R. M. Ortuño, Organometallics, 2007, 26, 3306 CrossRef CAS.
- M. P. Doyle, R. L. Dorrow and W. H. Tamblyn, J. Org. Chem., 1982, 47, 4059 CrossRef CAS.
- L. Jong and R. S. Stein, Macromolecules, 1991, 24, 2323 CrossRef CAS.
- D. R. Anton and R. H. Crabtree, Organometallics, 1983, 2, 855 CrossRef CAS.
- A. I. Olivos Suarez, J. N. H. Reek and B. de Bruin, J. Mol. Catal. A: Chem., 2010, 324, 24 CrossRef.
- M. Nagel and A. Sen, Organometallics, 2006, 25, 4722 CrossRef CAS.
- R. López-Fernandez, N. Carrera, A. C. Albéniz and P. Espinet, Organometallics, 2009, 28, 4996 CrossRef CAS.
- L. Ford and U. Jahn, Angew. Chem., Int. Ed., 2009, 48, 6386–6389 CrossRef CAS.
- S. Mecking, L. K. Johnson, L. Wang and M. Brookhart, J. Am. Chem. Soc., 1998, 120, 888 CrossRef CAS.
- G. Berthon-Gelloz, M. A. Siegler, A. L. Spek, B. Tinant, J. N. H. Reek and I. E. Markó, Dalton Trans., 2010, 39, 1444 RSC.
- H. van der Poel, G. van Koten and K. Vrieze, Inorg. Chem., 1980, 19, 1145 CrossRef CAS.
- L. K. Johnson, C. M. Killian and M. Brookhart, J. Am. Chem. Soc., 1995, 117, 6414 CrossRef CAS.
- R. van Asselt, E. E. C. G. Gielens, R. E. Rülke, K. Vrieze and C. J. Elsevier, J. Am. Chem. Soc., 1994, 116, 977 CrossRef CAS.
- R. E. Rülke, J. M. Ernsting, A. L. Spek, C. J. Elsevier, P. W. N. M. van Leeuwen and K. Vrieze, Inorg. Chem., 1993, 32, 5769 CrossRef CAS.
- B. Milani, A. Anzilutti, L. Vicentini, A. Sessanta o Santi, E. Zangrando, S. Geremia and G. Mestroni, Organometallics, 1997, 16, 5064 CrossRef CAS.
- J. W. Sprengers, J. Wassenaar, N. D. Clement, K. J. Cavell and C. J. Elsevier, Angew. Chem., Int. Ed., 2005, 44, 2026 CrossRef CAS.
Footnote |
| † This paper is part of a Polymer Chemistry issue highlighting the work of emerging investigators in the polymer chemistry field. Guest editors: Rachel O'Reilly and Andrew Dove. |
| This journal is © The Royal Society of Chemistry 2011 |