Tunable thermo-responsive polymer–protein conjugates via a combination of nucleophilic thiol–ene “click” and SET-LRP†
Mathew W.
Jones
a,
Matthew I.
Gibson
a,
Giuseppe
Mantovani
b and
David M.
Haddleton
a
aDepartment of Chemistry, University of Warwick, Coventry, UK CV4 7AL. E-mail: D.M.Haddleton@warwick.ac.uk
bSchool of Pharmacy, University of Nottingham, Nottingham, UK NG7 2RD
First published on 9th November 2010
Abstract
Herein we report the synthesis of a protein macroinitiator in a one-pot strategy using phosphine-mediated thiol–ene “click”, the macroinitiator was used to polymerise ethylene glycol containing monomers to yield polymer–protein conjugates with tunable thermoresponsive behaviour.
Conjugation of synthetic polymers to biological therapeutics is of considerable interest.1 Many biological properties of polypeptide-based drugs, such as stability, plasma half-lives and bioavailability, can be substantially improved following conjugation with an appropriate polymer.2–4Conjugation of poly(ethylene glycol) (PEG, PEGylation) is now a common strategy. Most initial PEGylations used lysine (primary amine) attachment, usually leading to statistical multi-site attachment.5
However, multi-site attachment leads to a mixture of products with consequences for bioactivity. In order to circumvent this, conjugation to other residues has been developed to introduce polymers at specific and well-defined sites. Cysteines have been explored as these are relatively rare, although biochemical modification can be utilised to introduce free thiols at specific positions. In addition disulfide bridges can be reduced to give two potential conjugation sites.6
Several groups have reported the synthesis of “smart” polymer conjugates, which respond to external stimuli such as temperature, pH or exposure to light/radiation.7,8Poly(N-isopropylacrylamide) (polyNIPAAm) has been studied due to its lower critical solution temperature (LCST) value of 32 °C, which confers temperature sensitivity to the conjugate.6,9,10
Homo- and co-polymers of ethylene glycol-based (macro)monomers have a number of potential biological applications including drug carriers11 and controlling surface cell adhesion12 and have been utilised to synthesise water-soluble polymers using controlled radical polymerisation techniques.13,14Polymers of the smaller ethylene glycol based monomers di(ethylene glycol) methyl ether methacrylate (DEGMEMA)15 (LCST ≈ 26 °C) and tri(ethylene glycol) methyl ether methacrylate (TEGMEMA) (LCST ≈ 52 °C)16 are excellent thermoresponsive materials.17Copolymerisation of these monomers gives polymers with tunable LCSTs.18,19 Two recent examples have emerged whereby thermoresponsive oligo(ethylene glycol)-based polymers have been successfully conjugated to proteins. Lutz et al. reported the copolymerisation of DEGMEMA with longer oligo(ethylene glycol) methacrylates from succinimidyl initiators, followed by conjugation to the lysine residues of trypsin. The obtained bioconjugate was not only thermoresponsive but also exhibited a higher enzymatic stability than native trypsin.20 Nolte et al. have also reported the polymerisation of discrete ethylene glycol based monomers by ATRP. Post-polymerisation modification yielded an azide-functional polymer, which was conjugated to alkyne-functional green fluorescent protein by CuAAC to yield the thermoresponsive bioconjugate.21
Highly efficient chemical transformations are essential in polymer chemistry to yield well defined materials.22Thiol–ene “click” chemistry23 has emerged as a valuable tool in polymer chemistry for star and dendrimer synthesis.24,25 This particular “click” reaction can proceed via two pathways: (i) a nucleophile or base catalysed Michael addition or (ii) an anti-Markovnikov radical addition. Both of these reactions are gaining considerable interest in recent literature.26,27 Single-electron transfer living radical polymerisation (SET-LRP) has been shown to give excellent control over molecular weight, molecular weight distributions and end group fidelity.28–30
The “grafting from” approach to polymer–protein conjugates has the advantage that isolation of the product involves the separation of a high molecular weight conjugate from a low molecular weight species.10,31 Herein, we report the utilisation of this technique for the synthesis of well-defined polymer–protein bioconjugates at ambient temperatures in polar solvents. This coupled with the ease of catalyst preparation and product recovery make SET-LRP an ideal system for this.
We have recently described the one-pot synthesis of polymer–protein conjugates, whereby a disulfide bridge was first reduced, using tris(2-carboxyethyl)phosphine (TCEP), a water-soluble phosphine, and subsequently conjugated to linear acrylic functional PEG chains using the same phosphine as a catalyst.32 Salmon calcitonin (sCT) is a 32 amino acid calcitropic hormone currently administered for the treatment of a number of hypercalcemia-related diseases and the disulfide bridge can be reduced whilst still retaining significant bioactivity. We envisaged that the same protocol could be used to introduce small-molecules into polypeptides. sCT was reduced in the presence of TCEP to give two thiols available for subsequent conjugation, followed by addition of ionomer (1) (Scheme 1). Conjugation was monitored by RP-HPLC and seemingly quantitative conversion of the reduced polypeptide to the bi-functionalised macroinitiator was observed after 60 minutes (Fig. 1). The macroinitiator was purified by dialysis and isolated as a white powder following lyophilisation. Analysis by MALDI-ToF-MS showed a well-defined peak corresponding to the difunctional polypeptide (Fig. 2), with a small peak corresponding to the loss of HBr.
 | ||
| Fig. 1 RP-HPLC analysis of the sCT macroinitiator. | ||
 | ||
| Fig. 2 MALDI-ToF-MS analysis of sCT macroinitiator. | ||
 | ||
| Scheme 1 Scheme 1One-pot synthetic protocol to sCT macroinitiator. | ||
Sacrificial initiators and solid phase initiators are useful in improving control by increasing the total initiator concentration in solution.6 Initial polymerisation studies were carried out using a soluble initiator (ethyl 2-bromoisobutyrate) and a solid-supported (Wang resin) initiator in order to assess the reaction timescales. DEGMEMA was polymerised by SET-LRP from both initiators in the presence of 10 mol% of Cu(II)Br2. Similar rates were observed, with living polymerisation characteristics and an absence of both an induction period and an exotherm,33 with >90% conversions in 4 hours (ESI†).
SET-LRP was subsequently used to polymerise DEGMEMA, TEGMEMA and an equimolar mixture of DEGMEMA/TEGMEMA in the presence of macroinitiator, 1, and the sacrificial initiator-functional Wang resin using identical reaction conditions (Scheme 2). SET-LRP was conducted at room temperature using DMSO as a solvent and 5 cm of copper wire as the heterogeneous catalyst. The polymerisation was quenched after 3 hours, so as to minimise any unwanted side-reactions such as bimolecular termination, which could result in bioconjugate cross-linking. The solution was immediately filtered to remove copper residues and insoluble Wang resin-supported polymer. The solution was dialysed extensively against water for 3 days to remove residual monomer and other small molecules. Lyophilisation of the solution yielded the conjugates as colourless oils. These were analysed by GPC and their LCSTs determined by turbidimetry, Table 1.
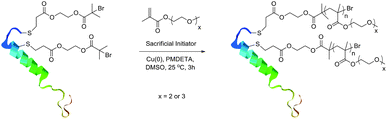 | ||
| Scheme 2 SET-LRP of discrete oligo(ethylene glycol) methacrylates from sCT macroinitiator. | ||
GPC with UV detection indicated the incorporation of the peptide in all of the polymers (ESI†), demonstrating successful ‘grafting from’ 1. The observed molecular weights were larger than expected, which is ascribed to a combination of the grafted architecture of the conjugate and the use of PMMA standards. All of the GPC traces (Fig. 3 and ESI†) showed a small high molecular weight shoulder, which according to the UV detector also contained protein, suggesting some solution aggregation had occurred.
 | ||
| Fig. 3 GPC trace of sCT–poly(DEGMEMA) conjugate. The predominant peak corresponds to the conjugate as well as observed higher molecular weight aggregated species. | ||
Polymerisations displayed linear 1st order kinetics (ESI†) implying a constant number of active polymer chains. This observation is not consistent with a high-molecular weight conjugate. Furthermore, the size of the shoulder appeared to be concentration-dependent indicating aggregation. Dynamic light scattering (DLS) analysis in DMF showed a peak at approximately 7 nm (ESI†) for each conjugate confirming a small amount of aggregation in the GPC solvent.
The LCSTs of the conjugates were evaluated by turbidimetry, Fig. 4. Upon heating, P1 showed a sharp transition at 24 °C and P2 at 51 °C. In order to demonstrate tunability P3 was analysed which contained a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of the two comonomers. P3 showed a single transition at 37 °C, indicating that the LCST can be fine-tuned by statistical copolymerisation.
:1 mixture of the two comonomers. P3 showed a single transition at 37 °C, indicating that the LCST can be fine-tuned by statistical copolymerisation.
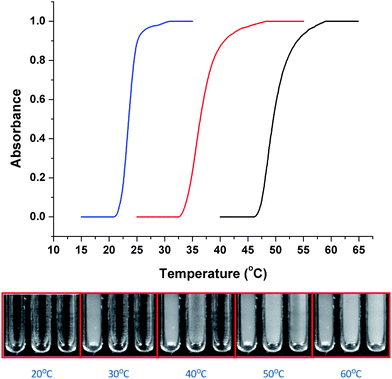 | ||
| Fig. 4 Cloud point analysis of sCT–polymer conjugates as a function of temperature. sCT–(DEGMEMA) (blue trace, left tube), sCT–(DEGMEMA-co-TEGMEMA) (red trace, middle tube), sCT–(TEGMEMA) (black trace, right tube). | ||
Thermoresponsive behaviour of these conjugates was further investigated by dynamic light scattering (DLS) at different temperatures. DLS is more sensitive to small changes in the conjugate structure than turbidimetry, which requires macromolecular precipitation. Upon heating P1 from 23 to 26 °C there was a dramatic increase in size from 7 to 620 nm indicating that upon heating through the LCST, there is a rapid transition which is essential for such conjugates to be employed for biomedical applications. Similar size increases were observed for the other conjugates, see ESI†.
In conclusion, SET-LRP has been successfully employed for the synthesis of tunable thermoresponsive protein–polymer conjugates. Discrete oligo(ethylene glycol) methacrylates were polymerised directly from a salmon calcitonin macroinitiator, 1, readily synthesised in a one-pot protocol utilising thiol–ene chemistry to yield well-defined conjugates. Conjugates with tunable cloud points ranging from 24 °C to 51 °C were synthesised using only DEGMEMA and TEGMEMA.
We thank EPSRC and Warwick Effect Polymers Ltd for funding; equipment used was supported by the Innovative Uses for Advanced Materials in the Modern World (AM2), with support from Advantage West Midlands (AWM) and part funded by the European Regional Development Fund (ERDF). MIG is a Birmingham Science City Interdisciplinary Research Fellow, supported HEFCE.
References
- R. Duncan, Nat. Rev. Drug Discovery, 2003, 2, 347–360 CrossRef CAS.
- S. M. Ryan, G. Mantovani, X. X. Wang, D. M. Haddleton and D. J. Brayden, Expert Opin. Drug Delivery, 2008, 5, 371–383 Search PubMed.
- B. Le Droumaguet and J. Nicolas, Polym. Chem., 2010, 1, 563–598 RSC.
- K. Velonia, Polym. Chem., 2010, 1, 944–952 RSC.
- J. Nicolas, G. Mantovani and D. M. Haddleton, Macromol. Rapid Commun., 2007, 28, 1083–1111 CrossRef CAS.
- K. L. Heredia, D. Bontempo, T. Ly, J. T. Byers, S. Halstenberg and H. D. Maynard, J. Am. Chem. Soc., 2005, 127, 16955–16960 CrossRef CAS.
- E. S. Gil and S. M. Hudson, Prog. Polym. Sci., 2004, 29, 1173–1222 CrossRef CAS.
- C. D. H. Alarcon, S. Pennadam and C. Alexander, Chem. Soc. Rev., 2005, 34, 276–285 RSC.
- M. Li, P. De, S. R. Gondi and B. S. Sumerlin, Macromol. Rapid Commun., 2008, 29, 1172–1176 CrossRef CAS.
- P. De, M. Li, S. R. Gondi and B. S. Sumerlin, J. Am. Chem. Soc., 2008, 130, 11288–11289 CrossRef CAS.
- G. Pasparakis and C. Alexander, Angew. Chem., Int. Ed., 2008, 47, 4847–4850 CrossRef CAS.
- E. Wischerhoff, K. Uhlig, A. Lankenau, H. G. Borner, A. Laschewsky, C. Duschl and J. F. Lutz, Angew. Chem., Int. Ed., 2008, 47, 5666–5668 CrossRef CAS.
- L. Tao, G. Mantovani, F. Lecolley and D. M. Haddleton, J. Am. Chem. Soc., 2004, 126, 13220–13221 CrossRef CAS.
- G. Mantovani, F. Lecolley, L. Tao, D. M. Haddleton, J. Clerx, J. J. L. M. Cornelissen and K. Velonia, J. Am. Chem. Soc., 2005, 127, 2966–2973 CrossRef CAS.
- S. Han, M. Hagiwara and T. Ishizone, Macromolecules, 2003, 36, 8312–8319 CrossRef CAS.
- T. Ishizone, A. Seki, M. Hagiwara, S. Han, H. Yokoyama, A. Oyane, A. Deffieux and S. Carlotti, Macromolecules, 2008, 41, 2963–2967 CrossRef CAS.
- J. F. Lutz, O. Akdemir and A. Hoth, J. Am. Chem. Soc., 2006, 128, 13046–13047 CrossRef CAS.
- J.-F. Lutz and A. Hoth, Macromolecules, 2006, 39, 893–896 CrossRef CAS.
- G. Chen, P. M. Wright, J. Geng, G. Mantovani and D. M. Haddleton, Chem. Commun., 2008, 1097–1099 RSC.
- Z. Zarafshani, T. Obata and J. F. Lutz, Biomacromolecules, 2010, 11, 2130–2135 CrossRef CAS.
- C. Lavigueur, J. G. Garcia, L. Hendriks, R. Hoogenboom, J. J. L. M. Cornelissen and R. J. M. Nolte, Polym. Chem., 2011 10.1039/c0py00229a.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- A. B. Lowe, Polym. Chem., 2010, 1, 17–36 RSC.
- J. W. Chan, B. Yu, C. E. Hoyle and A. B. Lowe, Chem. Commun., 2008, 4959–4961 RSC.
- K. L. Killops, L. M. Campos and C. J. Hawker, J. Am. Chem. Soc., 2008, 130, 5062–5064 CrossRef CAS.
- A. Dondoni, Angew. Chem., Int. Ed., 2008, 47, 8995–8997 CrossRef CAS.
- C. E. Hoyle, A. B. Lowe and C. N. Bowman, Chem. Soc. Rev., 2010, 39, 1355–1387 RSC.
- V. Percec, T. Guliashvili, J. S. Ladislaw, A. Wistrand, A. Stjerndahl, M. J. Sienkowska, M. J. Monteiro and S. Sahoo, J. Am. Chem. Soc., 2006, 128, 14156–14165 CrossRef CAS.
- G. Lligadas and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2745–2754 CrossRef CAS.
- N. H. Nguyen, B. M. Rosen, X. Jiang, S. Fleischmann and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5577–5590 CrossRef CAS.
- J. Nicolas, V. San Miguel, G. Mantovani and D. M. Haddleton, Chem. Commun., 2006, 4697–4699 RSC.
- M. W. Jones, G. Mantovani, S. M. Ryan, X. X. Wang, D. J. Brayden and D. M. Haddleton, Chem. Commun., 2009, 5272–5274 RSC.
- M. E. Levere, I. Willoughby, S. O'Donohue, A. de Cuendias, A. J. Grice, C. Fidge, C. R. Becer and D. M. Haddleton, Polym. Chem., 2010, 1, 1086–1094 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterisation of sCT conjugates. See DOI: 10.1039/c0py00329h |
| This journal is © The Royal Society of Chemistry 2011 |