Conjugated polyelectrolyte as signal amplifier for fluorogenic probe based enzyme activity study†
Ruoyu
Zhan
a,
Angela Jun Hiong
Tan
b and
Bin
Liu
*a
aDepartment of Chemical and Biomolecular Engineering, National University of Singapore, 4 Engineering Drive 4, Singapore, 117576. Fax: +65 67791936; Tel: +65 65168049
bDSO National Laboratories 20 Science Park Drive, Singapore, 118230. E-mail: cheliub@nus.edu.sg
First published on 2nd November 2010
Abstract
A cationic conjugated polyelectrolyte (CPE) is used as a signal amplifier to enhance the sensory response of a fluorogenic substrate (fluorescein diacetate) for esterase activity study. As fluorescein diacetate is nonfluorescent, it can be cleaved by esterase to yield a negatively charged fluorescent dye. Electrostatic interaction between the cationic CPE and negatively charged fluorescent dye leads to complexes which allow fluorescence resonance energy transfer to amplify the dye signal by a factor of up to ∼19-fold. The presence of CPE does not obviously influence the enzyme efficiency and the assay allows real-time monitoring of enzyme activity. As compared with common fluorogenic assays, the CPE amplified system has the advantage of shorter readout time with higher sensitivity, which could have wide applications in high throughput screening of enzyme activity.
Introduction
Esterase is a group of enzymes which catalyse the hydrolysis of fatty acid esters into acids and alcohols with water. Due to its broad substrate tolerance, esterase serves as a useful biocatalyst in industrial applications.1 A fluorometric method which utilizes fluorogenic probes as the substrate is one of the most commonly used methods for esterase activity studies.2 These assays rely on the development of fluorescence in solution as a result of substrate hydrolysis, which often has sensitivity in the micromolar to submicromolar range.3 To improve the assay sensitivity, substantial work has been focused on the development of more sensitive fluorescent substrate.4 In addition, fluorescence resonance energy transfer (FRET) based ratiometric probes containing donor and acceptor moieties linked via an ester linker have also been synthesized for esterase activity study.5 The cleavage of ester bond leads to the separation of FRET pairs and the reduced acceptor emission indicates the presence of enzyme in solution. Although these assays have been proven successful in esterase activity studies, further improvement in assay sensitivity at shorter readout times without assay complexicity is highly desirable.Conjugated polyelectrolytes (CPEs) are water-soluble π-conjugated polymers with charged side chain functionalities.6 Their electron delocalized backbone structure allows for effective electronic coupling and therefore fast intra- and interchain energy transfer.7CPEs have been used in various assays for chemical and biological target detection based on both superquenching and fluorescence resonance energy transfer (FRET) mechanisms.8 In addition, fluorescence-based enzymatic assays have been developed to monitor enzyme activities by taking advantage of the intrinsic fluorescent signal change of CPEs upon interaction with different substrates and enzyme digested products. One strategy relies on fluorescence quenching of CPEs upon complexation or covalent binding with quencher-labeled biomolecules and subsequent fluorescence change upon enzymatic hydrolysis of the substrate.9 The other strategy involves enzyme digestion-triggered changes in FRET between CPEs and organic dyes.10 These CPE-based enzymatic assays generally involve either modification of the substrate with fluorescent dye/quencher or pre-modification of CPE structures, which require multiple-step operations and the substrate modification may also lead to different kinetic factors and binding constants as compared to that for the nature substrate.
In this study, we take advantage of efficient energy transfer between CPEs and organic dye molecules to demonstrate that CPEs indeed could serve as signal amplifiers for fluorogenic probe based enzyme assays. As a proof of concept, we choose the most widely used fluorescein diacetate (FDA) as the substrate for esterase (pig liver esterase, PLE) activity study. We show that the assay sensitivity has improved about 19-fold in the presence of CPE, and the polymer does not obviously affect the enzyme efficiency.
Results and discussion
The chemical structures of the polymer and substrate and the schematic illustration of amplified fluorescence detection of enzyme activity are shown in Scheme 1. The FDA substrate is neutral and non fluorescent.11 In the absence of PLE, there is no electrostatic interaction between the CPE and FDA. No FRET occurs and only the polymer emission is detectable upon excitation of the CPE. After addition of PLE, FDA undergoes catalytic hydrolysis to yield fluorescent fluorescein (Fl) molecule, which has two negative charges at pH = 7.4.11,12 Electrostatic attraction between the CPE and anionic Fl brings them into close proximity for FRET, and the amplified signal facilitates enzyme activity study.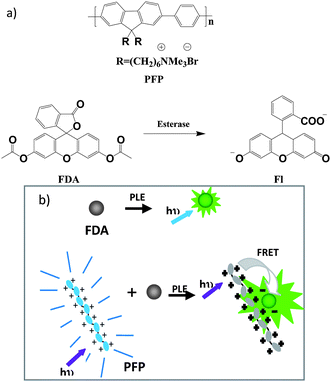 | ||
| Scheme 1 (a) Structures of the polymer, esterase substrate and hydrolyzed product. (b) A schematic illustration of fluorescence amplified detection of enzyme activity with a cationic CPE. | ||
FRET experiment
Poly[(9,9-bis(6′-N,N,N-trimethylammonium)-hexyl)fluorene-co-phenylene dibromide] (PFP, structure shown in Scheme 1) was chosen as the cationic light harvesting CPE for this study.13 The absorption and emission spectra of PFP and fluorescein are shown in Fig. 1. PFP has absorption and emission maxima at 420 nm and 490 nm, respectively. There is a good spectral overlap between the emission of PFP and the absorption of fluorescein, which should favor energy transfer between them. In addition, since the substrate FDA is neutral, no electrostatic interaction occurs between PFP and FDA, and the solution will have a very low background signal in the absence of enzyme.![Normalized absorption (dashed line) and PL spectra (solid line) of PFP (black) and Fl (red) in DMSO : PBS = 1 : 4 (v/v), pH = 7.4, [PBS] = 1 mM.](/image/article/2011/PY/c0py00265h/c0py00265h-f1.gif) | ||
Fig. 1 Normalized absorption (dashed line) and PL spectra (solid line) of PFP (black) and Fl (red) in DMSO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PBS = 1:4 (v/v), pH = 7.4, [PBS] = 1 mM. :PBS = 1:4 (v/v), pH = 7.4, [PBS] = 1 mM. | ||
According to the literature report, FDA could be fully hydrolyzed to Fl under very mild conditions. After 100 nM of FDA was hydrolyzed by 15.4 nM PLE in PBS buffer (1 mM, DMSO:PBS = 1:4 v/v, pH = 7.4), the solution fluorescence intensity was monitored until it reached saturation, which was almost the same as that for 100 nM Fl in the same buffer solution. PFP was subsequently added dropwise at an interval of 0.67 μM, and the corresponding photoluminescence (PL) spectra are shown in Fig. 2. The initial addition of PFP leads to a sharp increase in Fl emission, which is saturated at PFP repeat unit [RU] = 6.7 μM. Further addition of PFP does not induce any emission intensity change. The wavelength of the emission peaks slightly red shifted from 520 nm in the absence of PFP to 527 nm in the presence of PFP, mainly due to the change of polarity near Fl upon complex formation.14 Detailed comparison of Fl emission in the presence of PFP (excitation at 380 nm) against that upon direct excitation of the same amount of Fl at 488 nm in the absence of the polymer reveals that there is ∼19-fold increase in Fl signal, which indicates signal amplification provided by the light-harvesting PFP.
 | ||
| Fig. 2 Emission spectra of the solution containing 100 nM FDA upon PLE hydrolysis, followed by subsequent PFP addition at intervals of 0.67 μM. Direct excitation of Fl at 490 nm is also shown for comparison (black). | ||
PFP based assay
The PFP amplified esterase digestion experiments were conducted in PBS buffer (DMSO:PBS = 1:4 v/v, pH = 7.4, [PBS] = 1 mM) containing 100 nM FDA, 6.7 μM PFP and 15.4 nM PLE at 23 °C. Fig. 3 shows the fluorescence spectra change of PFP and FDA solution as a function of esterase digestion time. The initial emission of PFP and FDA in the absence of PLE shows intense blue emission at 420 nm from PFP. Upon addition of PLE and with increased esterase digestion time, the blue emission intensity of PFP at 420 nm gradually decreases over the incubating time from 0 to 30 min. Meanwhile, the emission of Fl at 527 nm shows a gradual increase over the full incubating time. This indicates that more esterase digested product F1 is formed and binds to PFP through electrostatic interaction to favor energy transfer. It is important to note the final Fl emission intensity upon polymer excitation is almost the same as that shown in Fig. 2, which indicates that the FDA has been fully hydrolyzed to Fl even in the presence of PFP.
![Time dependent emission spectra of PFP/FDA/PLE upon excitation at 380 nm. [FDA] = 100 nM, [PLE] = 15.4 nM, PFP [RU] = 6.7 μM; DMSO : PBS= 1 : 4 v/v, pH = 7.4, [PBS] = 1 mM.](/image/article/2011/PY/c0py00265h/c0py00265h-f3.gif) | ||
| Fig. 3 Time dependent emission spectra of PFP/FDA/PLE upon excitation at 380 nm. [FDA] = 100 nM, [PLE] = 15.4 nM, PFP [RU] = 6.7 μM; DMSO:PBS= 1:4 v/v, pH = 7.4, [PBS] = 1 mM. | ||
To demonstrate the application of PFP and FDA mixture in monitoring real-time esterase activity, the fluorescence intensity of F1 at 527 nm was studied as a function of esterase concentration. Fig. 4 shows the response of F1 intensity to esterase digestion reaction with esterase concentration varying from 0.0772 to 15.4 nM. The enlarged curves for esterase concentration from 0.0772 to 0.309 nM are shown in Fig. 5. At each esterase concentration, there is an increase in F1 intensity with increased incubation time. After incubation for 12 min, the F1 increases ∼80% of its final intensity for solutions containing 15.4 nM esterase, and the intensity reaches a plateau after 25 min. While in the presence of a lower concentration of esterase (e.g. 3.09 nM), the intensity reaches a quarter of that for 15.4 nM esterase in 12 min, and the emission intensity does not reach a plateau within 30 min. This observation indicates that increasing the esterase concentration gives rise to a higher cleavage reaction rate and less time is required to complete the digestion process.
![PL intensity at 527 nm as a function of time with various PLE concentrations. [FDA] = 100 nM, PFP [RU] = 6.7 μM, [PLE] = 15.4, 7.72, 3.09, 1.54, 0.772, 0.309, 0.154, 0.0772 nM. DMSO : PBS = 1 : 4 v/v, pH = 7.4, [PBS] = 1 mM. The polymer emission tail at 527 nm is subtracted.](/image/article/2011/PY/c0py00265h/c0py00265h-f4.gif) | ||
| Fig. 4
PL intensity at 527 nm as a function of time with various PLE concentrations. [FDA] = 100 nM, PFP [RU] = 6.7 μM, [PLE] = 15.4, 7.72, 3.09, 1.54, 0.772, 0.309, 0.154, 0.0772 nM. DMSO:PBS = 1:4 v/v, pH = 7.4, [PBS] = 1 mM. The polymer emission tail at 527 nm is subtracted. | ||
![PL intensity at 527 nm as a function of time for PFP/FDA/PLE solutions with various PLE concentrations. [FDA] = 100 nM, [PLE] = 0.309, 0.154, 0.0772 nM, PFP [RU] = 6.7 μM; DMSO : PBS = 1 : 4 v/v, pH = 7.4, [PBS] = 1 mM. Polymer emission tail is subtracted. The inset shows the PL intensity at 520 nm as a function of time for FDA/PLE solutions with various PLE concentrations in the absence of PFP. [FDA] = 100 nM, [PLE] = 1.54, 3.09 and 7.72 nM. DMSO : PBS = 1 : 4 v/v, pH = 7.4, [PBS] = 1 mM.](/image/article/2011/PY/c0py00265h/c0py00265h-f5.gif) | ||
| Fig. 5
PL intensity at 527 nm as a function of time for PFP/FDA/PLE solutions with various PLE concentrations. [FDA] = 100 nM, [PLE] = 0.309, 0.154, 0.0772 nM, PFP [RU] = 6.7 μM; DMSO:PBS = 1:4 v/v, pH = 7.4, [PBS] = 1 mM. Polymer emission tail is subtracted. The inset shows the PL intensity at 520 nm as a function of time for FDA/PLE solutions with various PLE concentrations in the absence of PFP. [FDA] = 100 nM, [PLE] = 1.54, 3.09 and 7.72 nM. DMSO:PBS = 1:4 v/v, pH = 7.4, [PBS] = 1 mM. | ||
To further demonstrate the advantage of PFP based assay, the fluorescence intensity at 520 nm as a function of esterase concentration in the absence of PFP was also studied. The inset of Fig. 5 shows the response of Fl intensity to esterase digestion reaction with PLE concentrations varying from 1.54 to 7.72 nM. At the same esterase concentration, the trend of the intensity curve in the presence and absence of PFP is similar, while the intensity is significantly amplified in the presence of PFP. After incubation for 30 min, the Fl intensity reached 6.5 a.u. for solution containing 1.54 nM PLE in the absence of PFP, while only ∼90 s is needed for solutions containing the same amount of esterase in the presence of PFP to reach the same intensity. As such, the improved signal output allows shorter read-out time for substrate digestion, which is beneficial to applications in high-throughput screening assays.
The selective response of the assay to esterase is demonstrated by incubating different hydrolases (2.5 μg mL−1) with 100 nM FDA in solutions containing 6.7 μM PFP RU for 30 min. The hydrolases include trypsin (Try), pepsin (Pep) and papain (Pap). Fig. 6 shows the PL intensity at 527 nm for each solution upon excitation at 380 nm. Only the solution containing PLE displays an intense fluorescence at 527 nm, and there is almost no detectable signal for solutions containing other enzymes (inset of Fig. 6). The control experiment using FDA alone also did not yield any signal at 527 nm upon excitation of the PFP. All these results indicate PFP based assay is selective towards PLE.
![PL intensity change at 527 nm of PFP/FDA/hydrolases upon excitation at 380 nm. [FDA] = 100 nM, [Try] = [Pep] = [Pap] = [PLE] = 2.5 μg mL−1, PFP [RU] = 6.7 μM; DMSO : PBS = 1 : 4 v/v, pH = 7.4, [PBS] = 1 mM. Incubation time is 30 min.](/image/article/2011/PY/c0py00265h/c0py00265h-f6.gif) | ||
| Fig. 6
PL intensity change at 527 nm of PFP/FDA/hydrolases upon excitation at 380 nm. [FDA] = 100 nM, [Try] = [Pep] = [Pap] = [PLE] = 2.5 μg mL−1, PFP [RU] = 6.7 μM; DMSO:PBS = 1:4 v/v, pH = 7.4, [PBS] = 1 mM. Incubation time is 30 min. | ||
Enzyme efficiency
To understand the influence of PFP on the esterase efficiency, the fluorescence intensity of Fl at 527 nm as a function of PFP addition time was studied. Fig. 7 shows the Fl intensity change upon addition of PFP solutions to FDA/PLE mixtures at different hydrolysis time of 0, 300, 600, 900, 1200 s. In these studies, the concentrations for PLE and PFP RU are 3.09 nM and 6.7 μM, respectively. Before addition of PFP, the solution fluorescence at 527 nm upon excitation at 380 nm is nearly zero. After addition of PFP, the solution fluorescence at 527 nm increases sharply, and all the curves for polymer addition at different time reach the same level of intensity. This result indicates that PFP only functions as a signal amplifier in the assay and it does not significantly affect the enzyme efficiency in hydrolysis of substrate.![PL intensity at 527 nm as a function of time with PFP added to PLE/FDA mixture at the hydrolysis time of 0, 300, 600, 900, 1200 s. [FDA] = 100 nM, [PLE] = 3.09 nM, PFP [RU] = 6.7 μM; DMSO : PBS = 1 : 4 v/v, pH = 7.4, [PBS] = 1 mM.](/image/article/2011/PY/c0py00265h/c0py00265h-f7.gif) | ||
| Fig. 7
PL intensity at 527 nm as a function of time with PFP added to PLE/FDA mixture at the hydrolysis time of 0, 300, 600, 900, 1200 s. [FDA] = 100 nM, [PLE] = 3.09 nM, PFP [RU] = 6.7 μM; DMSO:PBS = 1:4 v/v, pH = 7.4, [PBS] = 1 mM. | ||
Conclusion
In summary, we show that PFP could be used as a signal amplifier to improve the sensitivity of commercially available fluorogenic assays by taking advantage of the light-harvesting property of PFP and efficient FRET to the hydrolyzed product. Real time detection of enzyme activity is demonstrated and the presence of PFP is found not to have any obvious influence on the enzyme efficiency. Compared with common fluorescent assays, the PFP amplified strategy has the advantage of shorter readout time and higher sensitivity, which is highly beneficial for fast screening of enzyme inhibitors and studying of enzyme activity.Experiments
Materials
All chemicals were purchased from Sigma-Aldrich unless otherwise noted. Pig liver esterase (PLE) from Fluka has 131 U/mg, and its molecular weight is 162,000 Da. Poly[(9,9-bis(6′-N,N,N-trimethylammonium)-hexyl)fluorene-co-phenylene dibromide] (PFP) was synthesized according to the literature. Stock solutions of PLE (9.24 μM) in water, fluorescein diacetate (FDA, 50 μM) in DMSO were prepared and stored at −20 °C. A stock solution of PFP (1 mM) in water was prepared based on the repeat unit (RU) and was stored at 4 °C. Milli-Q water (18.2 MΩ) was used for experiments.Instruments
Fluorescence emission spectra were recorded on a Perkin-Elmer LS-55 equipped with a xenon lamp excitation source and a Hamamatsu (Japan) 928 PMT, using 90° angle detection for solution samples. The excitation energy at different wavelength was automatically adjusted to the same level by an excitation correction file. The instrument was controlled by FL WinLab Software. Detections were conducted under single scan, kinetic scan and multiple-channel time dependent luminescence modes. Biokinetic accessory was used and the speed of the magnetic stir was low.Detection procedure
To evaluate the specific assay response to esterase, hydrolase stock solution was transferred to a 3 mL cuvette with a magnetic stir, a mixture of 6 μL FDA stock solution (50 μM), 20 μL PFP stock solution (1 mM), 600 μL DMSO and 2400 μL PBS buffer (pH = 7.4, [PBS] = 1 mM) was then added to the cuvette to yield the final [Hydrolase] = 2.5 μg mL−1, [FDA] = 100 nM and [PFP] = 6.7 μM. The spectra were collected at time 0 and 30 min, and in the range of 400–650 nm upon excitation of PFP at 380 nm.
Acknowledgements
The authors are grateful to the National University of Singapore (R-279-000-234-123), Singapore Ministry of Education (R-279-000-255-112) and Temasek Defense Systems Institute (R-279-000-268-422, R-279-000-268-592, and R-279-000-268-232) for financial support.Notes and references
- (a) Z. B. Liu, R. Weis and A. Glieder, Food Technol. Biotechnol., 2004, 42, 237–249 Search PubMed; (b) D. de Maria, C. A. Garcia-Burgos, G. Bargeman and R. W. van Gemert, Synthesis, 2007, 1439–1452 CrossRef.
- J. L. Reymond, Enzyme Assays: High-throughput Screening, Genetic Selection and Fingerprinting, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2006 Search PubMed.
- (a) D. N. Kramer and G. G. Guilbault, Anal. Chem., 1963, 35, 588–589 CrossRef CAS; (b) G. G. Guilbault and D. N. Kramer, Anal. Chem., 1964, 36, 409–412 CrossRef CAS.
- (a) Y. Z. Yang, P. Babiak and J. L. Reymond, Helv. Chim. Acta, 2006, 89, 404–415 CrossRef; (b) F. Y. Ge, L. G. Chen, X. L. Zhou, H. Y. Pan, F. Y. Yan, G. Y. Bai and X. L. Yan, Dyes Pigm., 2007, 72, 322–326 CrossRef CAS.
- (a) J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 3rd edn, 2006 Search PubMed; (b) Y. Z. Yang, P. Babiak and J. L. Reymond, Org. Biomol. Chem., 2006, 4, 1746–1754 RSC; (c) L. Yi, L. Cao, L. L. Liu and Z. Xi, Tetrahedron, 2008, 64, 8947–8951 CrossRef CAS.
- (a) M. R. Pinto and K. S. Schanze, Synthesis, 2002, 1293–1309 CrossRef CAS; (b) B. Liu and G. C. Bazan, Chem. Mater., 2004, 16, 4467–4476 CrossRef CAS.
- S. W. Thomas, G. D. Joly and T. M. Swager, Chem. Rev., 2007, 107, 1339–1386 CrossRef CAS.
- (a) H. A. Ho, A. Najari and M. Leclerc, Acc. Chem. Res., 2008, 41, 168–178 CrossRef CAS; (b) H. Jiang, P. Taranekar, J. R. Reynolds and K. Schanze, Angew. Chem., Int. Ed., 2009, 48, 4300–4316 CrossRef CAS; (c) K. Li and B. Liu, Polym. Chem., 2010, 1, 252–259 RSC; (d) K. Y. Pu and B. Liu, Biosens. Bioelectron., 2009, 24, 1067–1073 CrossRef CAS; (e) J. Z. Liu, Y. C. Zhong, P. Lu, Y. N. Hong, J. W. Y. Lam, M. Faisal, Y. Yu, K. S. Wong and B. Z. Tang, Polym. Chem., 2010, 1, 426–429 RSC.
- (a) S. Kumaraswamy, T. Bergstedt, X. B. Shi, F. Rininsland, S. Kushon, W. S. Xia, K. Ley, K. Achyuthan, D. McBranch and D. Whitten, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 7511–7515 CrossRef CAS; (b) F. Rininsland, W. S. Xia, S. Wittenburg, X. B. Shi, C. Stankewicz, K. Achyuthan, D. McBranch and D. Whitten, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 15295–15300 CrossRef CAS; (c) J. H. Wosnick, C. M. Mello and T. M. Swager, J. Am. Chem. Soc., 2005, 127, 3400–3405 CrossRef CAS.
- (a) F. D. Feng, F. He, L. L. An, S. Wang, Y. L. Li and D. B. Zhu, Adv. Mater., 2008, 20, 2959–2964 CrossRef CAS; (b) Y. Zhang, Y. Y. Wang and B. Liu, Anal. Chem., 2009, 81, 3731–3737 CrossRef CAS.
- G. Adam and H. Duncan, Soil Biol. Biochem., 2001, 33, 943–951 CrossRef CAS.
- L. L. Wang, A. Roitberg, C. Meuse and A. K. Gaigalas, Spectrochim. Acta, Part A, 2001, 57, 1781–1791 CrossRef CAS.
- B. Liu and G. C. Bazan, J. Am. Chem. Soc., 2006, 128, 1188–1196 CrossRef CAS.
- B. Liu, B. S. Gaylord, S. Wang and G. C. Bazan, J. Am. Chem. Soc., 2003, 125, 6705–6714 CrossRef CAS.
Footnote |
| † This paper is part of a Polymer Chemistry issue highlighting the work of emerging investigators in the polymer chemistry field. Guest Editors: Rachel O'Reilly and Andrew Dove. |
| This journal is © The Royal Society of Chemistry 2011 |