Supramolecular complexes of single walled carbon nanotubes with conjugated polymers†‡
Patigul
Imin
,
Fuyong
Cheng
and
Alex
Adronov
*
Department of Chemistry and Chemical Biology, The Brockhouse Institute for Materials Research, McMaster University, 1280 Main St W, Hamilton, ON L8S 4M1, Canada. E-mail: adronov@mcmaster.ca; Fax: +1(905) 521-2773; Tel: +1(905) 525-9140 ext. 23514
First published on 29th October 2010
Abstract
We have synthesized a series of conjugated polymers, including poly[(2,7-(9,9-dioctylfluorene)-alt-2,7-(9,9-didodecylfluorene)] (PF), poly[(2,7-(9,9-dioctylfluorene)-alt-2,5-(3-dodecylthiophene)] (PFT), and poly(3-dodecylthiophene) (PT). Non-covalent functionalization of single walled carbon nanotubes (SWNTs) with these polymers can impart good solubility to nanotubes in a number of organic solvents, including THF, dichlorobenzene, chloroform, and toluene. Solution and solid-state characterization of the resulting polymer–SWNT composites are described, including UV-Vis absorption and Raman spectroscopy. It was found that the UV-Vis absorption maximum of the polymers was red-shifted in their corresponding composites due to the planarization of the polymer backbone following adsorption upon the SWNT surface. Polymer–SWNT complexes also exhibited good solution stability at elevated temperature in THF and dichlorobenzene, with no significant SWNT sedimentation observed at elevated temperatures. Both UV-Vis absorption and Raman spectroscopy results indicated that the interaction of PT with the nanotubes was different from those of PF and PFT, suggesting that the choice of aromatic ring in the polymer structures plays an important role in the supramolecular complex formation with carbon nanotubes.
Introduction
Since the seminal work of Iijima,1carbon nanotubes (CNTs) have attracted tremendous research interest due to their extraordinary mechanical, electronic, and optical properties, and have become the most extensively studied materials in the field of nanotechnology.2,3 Although the favourable properties of CNTs make them potentially useful for a number of applications, their commercial exploitation has been limited due to difficulties related to their purification, dissolution, and solution-phase processing. To overcome their inherent insolubility and improve the processability of carbon nanotubes, it is desirable to functionalize them so as to generate nanotube derivatives that are compatible with common organic solvents as well as organic matrix materials.4,5 To this end, a variety of CNT surface modification techniques have been developed during the past few years, including methods for covalent6–9 and non-covalent functionalization.10–16Although the covalent functionalization strategy has proven effective in solubilizing both single-walled and multi-walled carbon nanotubes (SWNTs and MWNTs, respectively) in various organic solvents, as well as in water, the covalent linkage of functional groups to the nanotube surface results in the disruption of the sp2-hybridized carbon framework.17,18 This leads to a significant reduction in the electrical conductivity and mechanical strength of such chemically functionalized nanotubes, when compared to pristine CNTs.5,19,20 Conversely, when non-covalent functionalization is used, the structural integrity of carbon nanotubes remains unchanged, allowing retention of the CNT properties, and enabling the use of both their conductive and mechanical properties in eventual applications. It has been shown that various conjugated structures, such as pyrene,19,21,22porphyrins,12–14 and π-conjugated polymers10,23–42 can form supramolecular complexes with nanotubes, and the resulting complex materials exhibit good solubility. In recent years, composite materials based on conjugated polymers and SWNTs have generated significant interest due to the combination of optical, electrical, and processability properties of the conjugated polymers with the strength and conductivity properties of carbon nanotubes. A number of conjugated polymers, including poly(m-phenylene vinylene) (PmPV),23–27 poly(thiophene) (PT),28–35 poly(phenylene ethynylene) (PPE),10,41,42 and poly(fluorene)36–40 have been successfully used to modify and strongly interact with the carbon nanotube surface through π-stacking or helical wrapping.
Poly(fluorene), poly(thiophene), and derivatives of these polymers are among the most widely investigated conjugated polymers due to their excellent solubility, high thermal, photochemical, and environmental stability, and ease of control over their optoelectronic properties via macromolecular engineering.43–50 Recently, it has been found that fluorene-containing conjugated polymers can form stable complexes with SWNTs, and exhibit excellent solubility and solution-stability properties even after the removal of excess polymer.37 Interest in this class of conjugated polymers is increasing further as reports of selective solubilization of certain SWNT species have begun to be published.36,38–40 It is therefore desirable to develop a detailed understanding of how variations in the backbone of a conjugated polymer can affect the interactions between the polymer and the SWNT surface. However, few comparisons of different conjugated polymer backbones in terms of their supramolecular functionalization of CNTs have been reported.
Herein, we compare three different polymer backbones with respect to their interactions with SWNTs, including poly[(2,7-(9,9-dioctylfluorene)-alt-2,7-(9,9-didodecylfluorene)] (PF), poly(3-dodecyl thiophene) (PT), and an alternating copolymer of fluorene and thiophene, poly[(2,7-(9,9-dioctylfluorene)-alt-2,5-(3-dodecylthiophene)] (PFT). Specifically, the effect of changing the polymer backbone on the nanotube solubility, solution stability, and photophysical properties was investigated.
Results and discussion
Scheme 1 illustrates the synthesis of the three polymers, PF, PFT, and PT. The preparation of both PF and PFT involved Suzuki polycondensation of commercially available diborate (1) and the corresponding dibromides (2 and 3). Synthesis of PT was accomplished by a chemical oxidation method, using 3-dodecylthiophene as depicted in Scheme 1.44,45,49 The resulting polymers were soluble in common organic solvents, such as THF, chloroform, toluene, and dichlorobenzene. The polymer molecular weight and polydispersity index (PDI) values were estimated from gel permeation chromatography (GPC), using polystyrene standards, and are listed in Table 1. The 1H NMR spectra of the polymers were also in good agreement with the proposed polymer structures (see ESI†).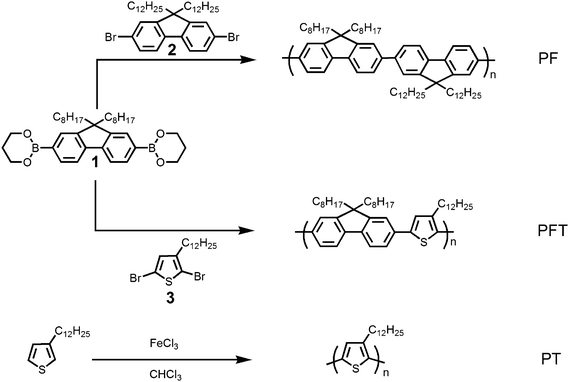 | ||
| Scheme 1 Synthesis of π-conjugated polymers PF, PFT and PT. | ||
| Polymer | M n/kg mol−1 | PDI |
|---|---|---|
| PF | 9 | 1.6 |
| PFT | 15 | 2.7 |
| PT | 48 | 1.5 |
To investigate the supramolecular interactions of these polymers with SWNTs, each polymer/nanotube mixture was prepared and treated identically. In a typical experiment, 20 mL of THF were added to a vial containing 10 mg SWNT and 30 mg conjugated polymer, and the mixture was sonicated for 1 h. The vial was centrifuged at 2576g for 20 min, and the resulting suspension was allowed to stand overnight. The dark-colored, clear supernatant was carefully transferred to another vial using a pipette. Then the isolated supernatant was further diluted with 50 mL of THF, sonicated for 5 min, filtered through a 200 nm pore diameter Teflon membrane, and repeatedly washed with THF until the filtrate was colorless and showed practically no fluorescence when irradiated using a UV lamp. 10 mL of THF were added to the recovered SWNT residue, and the vial was further sonicated for 5 min. The resulting dark suspension was centrifuged at 2576g for 20 min and allowed to stand overnight undisturbed. The supernatant was carefully transferred by a pipette to another vial, producing a solution of discrete polymer–nanotube complexes that have been separated from any unbound polymer. Fig. 1B–D show photographs of the conjugated polymer–SWNT complexes in THF, along with a comparison to pristine SWNTs treated in the same way (except for initial addition of the polymer) in THF. It was found that when using these fluorene and thiophene-based conjugated polymers, it was possible to produce homogeneous solutions in THF even after the removal of the excess unbound polymer, and the solutions were stable for at least four weeks without SWNT precipitation. Conversely, pristine SWNTs in the absence of polymers do not dissolve to any extent after similar treatment. Additionally, polymer–SWNT complexes exhibited good film-forming properties, easily producing uniform films by drop casting or spin coating at room temperature.
 | ||
| Fig. 1 Photograph of four samples in THF: A) pristine SWNT; B) PF–SWNT; C) PFT–SWNT; D) PT–SWNT. | ||
Photophysical properties of the polymer and polymer–nanotube complexes were investigated by steady-state UV-Vis spectroscopy. PF, PFT, and PT were found to exhibit absorption maxima at 380 nm, 403 nm, and 435 nm in THF solution, respectively, as shown in Fig. 2A(i). These absorption spectra are in agreement with previously reported values for similar polymers of nearly equal molecular weight.51–53 It is important to note that these three polymers represent a series in which the band gap systematically decreases upon the introduction of the electron rich and sterically less demanding thiophene units. The effect of this systematic variation to polymer composition and electronic structure on the supramolecular interaction of these fluorene and thiophene containing polymers with SWNTs in THF solution was initially evident from the UV-Vis absorption spectra of the polymer–nanotube complexes prepared as described above. From these spectra, depicted in Fig. 2A(ii), it is clear that the absorption characteristics of both the polymer and SWNTs are observable, even though the free polymer in solution was removed by extensive washing. In each case, it was found that the absorption maximum of the polymer exhibited a characteristic bathochromic shift once bound to the nanotube surface. This shift arises from an increased effective conjugation length within the polymer, most likely caused by the planarization of its backbone following adsorption upon the SWNT surface. It was also found that the magnitude of the red-shift varied with the polymer structure, as exhibited in Fig. 2A(ii). The largest absorption red-shift was observed in the PT–SWNT complex (∼120 nm) in solution, while a very small red-shift (∼4 nm) was observed when PF was used (Table 2).
 | ||
| Fig. 2 UV-Vis absorption spectra of the polymers (i) and polymer–nanotube complexes (ii) in THF (A), and as films on glass (B). | ||
| Polymers | Polymer in THF/nm | Polymer film/nm | Polymer–SWNT in THF/nm | Polymer–SWNT film/nm | Δ1a/nm | Δ2a/nm |
|---|---|---|---|---|---|---|
| a Note: Δ1 = UVλmax(polymer film) − UVλmax(polymer solution), Δ2 = UVλmax(polymer–SWNT solution) − UVλmax(polymer solution). | ||||||
| PF | 380 | 380 | 384 | 384 | 0 | 4 |
| PFT | 403 | 416 | 423 | 423 | 13 | 20 |
| PT | 435 | 509 | 558 | 558 | 74 | 123 |
It is known that poly(alkylthiophene)s adopt a poorly conjugated random coil structure in good solvents, and undergo a coil-to-rod transition when transferred from solution to the solid state, producing extended chains of coplanar thiophene rings.52,54 The large shift in PT–SWNT complex absorption indicates that, in our case, the PT also adopts a random coil conformation, and transitions to a much more planar structure when adsorbed onto the nanotube surface. This may also be the result of a very strong π-stacking interaction between the poly(thiophene) backbone and the nanotube surface. The smaller red shift for PF–SWNT and PFT–SWNT composites suggests that these polymers adopt a more rigid rod conformation in THF, and only slightly planarize once adsorbed onto the nanotube surface.
The preparation and characterization of high-quality, homogeneous thin films from conjugated polymers as well as nanotube composite materials are important for the fabrication of a wide variety of devices.45,55–57 Transparent and uniform films of PF, PFT, and PT, as well as the corresponding polymer–SWNT complex materials were prepared on glass slides by drop-casting from their THF solutions at room temperature. UV-Vis absorption spectra of the conjugated polymer and polymer–SWNT complex films are provided in Fig. 2B(i and ii). In the absorption spectra of the PF film, there was no red-shift observed relative to the corresponding solution spectrum. This indicates that there are no noticeable molecular conformation changes for PF when going from solution to solid state films. However, in PFT and PT films, significant bathochromic shifts (13 nm and 74 nm, respectively) were observed when compared to the solution-phase absorption spectra. This indicates that the polymers adopt more planar conformations when cast into thin films relative to their state in solution. Interestingly the magnitude of the red-shift for each polymer in the solid state is comparable to their corresponding complexes with the SWNTs in solution. Polymer–SWNT complex absorption spectra in the solid state exhibited absorption maxima that were similar to the corresponding polymer–SWNT complexes in solution (Fig. 2B(ii)).
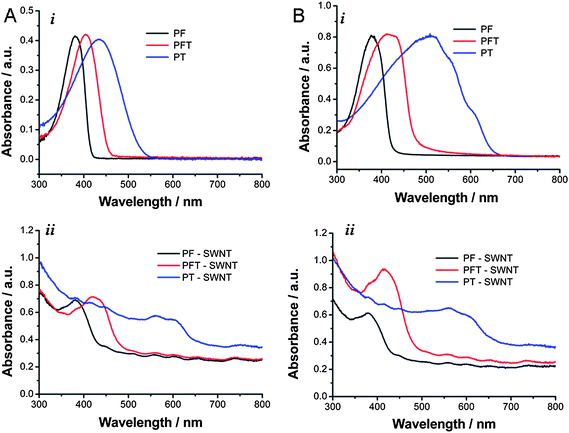
Solubility and solution stability of the conjugated polymer–SWNT complexes in other organic solvents, such as CHCl3, dichlorobenzene, and toluene, were also investigated in detail. In a typical experiment, 5 mL of solvent were added to a vial containing 2.5 mg SWNT and 2.5 mg conjugated polymer, and the mixture was sonicated for 30 min. The vial was allowed to stand one week, and then the dark-colored, clear supernatant was carefully transferred to another vial using a pipette. Previous experimental and theoretical studies showed that solvents with high density and polarity are better suited for SWNTs.58–60 Similarly, the present series of conjugated polymer–SWNT complexes exhibited very good solubility and solution stability in CHCl3 and dichlorobenzene (solvents with higher density), for periods of at least 3 months, with no observable precipitation. Quantitative flocculation and precipitation of the polymer–SWNT complexes in toluene were observed to slowly occur over several days. In addition, diluted solutions were prepared from each polymer–SWNT complex in different solvents, and the solution stability of the polymer–SWNT complexes at different temperatures was investigated by UV-Vis spectroscopy. Fig. 3 represents variable-temperature UV-vis absorption spectra of PF–SWNT (Fig. 3A), PFT–SWNT (Fig. 3B), and PT–SWNT (Fig. 3C) complexes in THF (i), dichlorobenzene (ii) and toluene (iii).
 | ||
| Fig. 3 UV-Vis absorption data of the polymer–nanotube complexes in THF, dichlorobenzene, and toluene at different temperatures. PF–SWNT (A), PFT–SWNT (B), and PT–SWNT (C) complexes in THF (i), dichlorobenzene (ii), and toluene (iii). | ||
Upon stepwise heating from room temperature to 65 °C in THF and up to 95 °C in dichlorobenzene, there was no visible carbon nanotube precipitation observed. However, the polymer–SWNT solution in toluene underwent significant precipitation during the course of the experiment, resulting in the light scattering and a noisy UV-Vis absorption spectrum. The absorption maxima of the polymers within polymer–SWNT complexes gradually decreased in intensity and slightly blue shifted with increasing temperature. This change in the absorption is mainly due to the conformational change of the polymer from a more planar conformation to relatively disordered states at higher temperature.43 The identical effect was observed for the polymers alone, in the absence of SWNTs (see ESI, Fig. S1†).
Raman spectroscopy was also used to further characterize the polymer–SWNT samples. Sample preparation involved drop casting the diluted polymer–SWNT solutions in THF onto a glass microscope slide, and drying in air prior to measurement. Raman spectra were collected at an excitation wavelength of 785 nm. The Raman spectra of pristine SWNTs (as received), and soluble polymer–SWNT complexes are shown in Fig. 4A, where all spectra are normalized to the graphitic (G) band (1590 cm−1). The position of the G band at ∼1590 cm−1 in polymer–SWNT complexes was almost identical to that of pristine nanotubes. Both the disorder (D) band at ∼1300 cm−1 and the second-order G′ band at 2600 cm−1 did not significantly increase in intensity after supramolecular functionalization. These results indicate that noncovalent functionalization of nanotubes with these conjugated polymers does not introduce any defects in nanotube electronic structures. The frequency and intensity of radial breathing mode (RBM) features in the range of 120–350 cm−1 provide the most valuable information for sample characterization.61,62 SWNT diameter distribution for the HiPco-produced nanotubes used in this study can be estimated according to the relationship between RBM frequency and tube diameter, νRBM = A/dt + B (where dt is the tube diameter in nm, νRBM is the Raman shift of the peak in cm−1, A is 223.5 cm−1 and B is 12.5 cm−1 for HiPco SWNTs).63 RBM profiles of the as-received HiPco sample as well as the soluble polymer–SWNT complexes are shown in Fig. 4B.
 | ||
| Fig. 4 Raman spectra of the polymer–nanotube complexes, polymer–SWNT soluble fraction at 785 nm excitation, showing (A) the entire spectral range, and (B) the RBM region. | ||
Comparing the RBM frequency and intensity, it was observed that the polymers show a preference for specific tube diameters in THF solution. In the spectrum of PF–SWNT and PFT–SWNT, the signals at 231 cm−1 and 224 cm−1 (corresponding to diameters of ∼1.02 and ∼1.06 nm respectively) showed much higher intensity, and were significantly narrower, when compared to the original SWNT sample, while the peak at ∼204 cm−1 (corresponding to a nanotube diameter of ∼1.17 nm) remained almost unchanged. The signal intensity at ∼265 cm−1 was decreased 2-fold for both PF–SWNT and PFT–SWNT when compared to the original pristine nanotube sample. Surprisingly, the RBM signal intensity is significantly different for the PT–SWNT sample when compared to the PF–SWNT and PFT–SWNT samples. A decrease in intensity in the signals at 231 cm−1 and 224 cm−1 for PT–SWNT was observed, while the peak at 204 cm−1 (corresponding to a nanotube diameter of ∼1.17 nm) is substantially increased in intensity. It is interesting to note that the PT–SWNT sample also showed the most profound difference in the UV-Vis measurements. These results seem to indicate that the relatively electron-rich PT may interact with the SWNTs in a different manner from the other two polymers, which may be the result of complementarity with relatively electron-poor semiconducting SWNTs that dominate the nanotube sample. Further studies of this effect are needed in order to fully understand this phenomenon.
Conclusion
A series of fluorene and thiophene containing conjugated polymers was synthesized and subsequently utilized for the preparation of supramolecular polymer–SWNT composite materials. It was demonstrated that the supramolecular interaction between SWNTs and PF, PFT, and PT polymers resulted in excellent solubility of the nanotubes in THF, even after the removal of the excess polymer. The resulting conjugated polymer–SWNT supramolecular complexes also exhibited solution stability at elevated temperature in several organic solvents, including THF, chloroform, and dichlorobenzene. The spectroscopic characterization of the polymer–SWNT complex materials indicated that the interaction between the conjugated polymers and SWNTs is influenced by the polymer structure. Specifically, the relatively electron-rich PT sample seems to exhibit a different interaction with the SWNT surface than the PF or PFT structures.Acknowledgements
Financial support for this work was provided by the Natural Science and Engineering Research Council of Canada (NSERC), Canada Foundation for Innovation (CFI), and Ontario Innovation Trust (OIT). PI gratefully acknowledges the Ontario Ministry of Training for an Ontario Graduate Scholarship.References
- S. Iijima, Nature, 1991, 354, 56–58 CrossRef CAS.
- D. A. Walters, L. M. Ericson, M. J. Casavant, J. Liu, D. T. Colbert, K. A. Smith and R. E. Smalley, Appl. Phys. Lett., 1999, 74, 3803–3805 CrossRef CAS.
- S. J. Tans, M. H. Devoret, H. J. Dai, A. Thess, R. E. Smalley, L. J. Geerligs and C. Dekker, Nature, 1997, 386, 474–477 CrossRef CAS.
- D. A. Britz and A. N. Khlobystov, Chem. Soc. Rev., 2006, 35, 637–659 RSC.
- A. Hirsch, Angew. Chem., Int. Ed., 2002, 41, 1853–1859 CrossRef CAS.
- K. Balasubramanian and M. Burghard, Small, 2005, 1, 180–192 CrossRef CAS.
- P. Liu, Eur. Polym. J., 2005, 41, 2693–2703 CrossRef CAS.
- F. Y. Cheng and A. Adronov, Chem. Mater., 2006, 18, 5389–5391 CrossRef CAS.
- C. M. Homenick, U. Sivasubramaniam and A. Adronov, Polym. Int., 2008, 57, 1007–1011 CrossRef CAS.
- J. Chen, H. Y. Liu, W. A. Weimer, M. D. Halls, D. H. Waldeck and G. C. Walker, J. Am. Chem. Soc., 2002, 124, 9034–9035 CrossRef CAS.
- G. J. Bahun, C. Wang and A. Adronov, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 1941–1951 CrossRef CAS.
- F. Y. Cheng, S. Zhang, A. Adronov, L. Echegoyen and F. Diederich, Chem.–Eur. J., 2006, 12, 6062–6070 CrossRef CAS.
- F. Y. Cheng and A. Adronov, Chem.–Eur. J., 2006, 12, 5053–5059 CrossRef CAS.
- F. Y. Cheng and A. Adronov, J. Porphyrins Phthalocyanines, 2007, 11, 198–204 CrossRef CAS.
- S. Lefrant, M. Baibarac, I. Baltog, J. Y. Mevellec, C. Godon and O. Chauvet, Diamond Relat. Mater., 2005, 14, 867–872 CrossRef.
- T. Casagrande, P. Imin, F. Y. Cheng, G. A. Botton, I. Zhitomirsky and A. Adronov, Chem. Mater., 2010, 22, 2741–2749 CrossRef CAS.
- A. Garg and S. B. Sinnott, Chem. Phys. Lett., 1998, 295, 273–278 CrossRef CAS.
- E. T. Mickelson, C. B. Huffman, A. G. Rinzler, R. E. Smalley, R. H. Hauge and J. L. Margrave, Chem. Phys. Lett., 1998, 296, 188–194 CrossRef CAS.
- N. Nakashima, Y. Tomonari and H. Murakami, Chem. Lett., 2002, 638–639 CrossRef CAS.
- M. S. Strano, C. A. Dyke, M. L. Usrey, P. W. Barone, M. J. Allen, H. W. Shan, C. Kittrell, R. H. Hauge, J. M. Tour and R. E. Smalley, Science, 2003, 301, 1519–1522 CrossRef CAS.
- R. J. Chen, Y. G. Zhang, D. W. Wang and H. J. Dai, J. Am. Chem. Soc., 2001, 123, 3838–3839 CrossRef CAS.
- P. Petrov, F. Stassin, C. Pagnoulle and R. Jerome, Chem. Commun., 2003, 2904–2905 RSC.
- T. Umeyama, N. Kadota, N. Tezuka, Y. Matano and H. Imahori, Chem. Phys. Lett., 2007, 444, 263–267 CrossRef CAS.
- S. M. Keogh, T. G. Hedderman, P. Lynch, G. F. Farrell and H. J. Byrne, J. Phys. Chem. B, 2006, 110, 19369–19374 CrossRef CAS.
- S. M. Keogh, T. G. Hedderman, E. Gregan, G. Farrell, G. Chambers and H. J. Byrne, J. Phys. Chem. B, 2004, 108, 6233–6241 CrossRef CAS.
- F. Massuyeau, H. Aarab, L. Mihut, S. Lefrant, E. Faulques, J. Wery, E. Mulazzi and R. Perego, J. Phys. Chem. C, 2007, 111, 15111–15118 CrossRef CAS.
- S. S. Chu, W. H. Yi, S. F. Wang, F. M. Li, W. K. Feng and Q. H. Gong, Chem. Phys. Lett., 2008, 451, 116–120 CrossRef CAS.
- A. Ikeda, K. Nobusawa, T. Hamano and J. Kikuchi, Org. Lett., 2006, 8, 5489–5492 CrossRef CAS.
- M. R. Karim, C. J. Lee and M. S. Lee, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 5283–5290 CrossRef CAS.
- J. Tsukamoto and J. Mata, Jpn. J. Appl. Phys., Part 2, 2004, 43, 214–216.
- R. G. S. Goh, J. M. Bell, N. Motta and E. R. Waclawik, J. Nanosci. Nanotechnol., 2006, 6, 3929–3933 CrossRef CAS.
- R. G. S. Goh, N. Motta, J. M. Bell and E. R. Waclawik, Appl. Phys. Lett., 2006, 88, 053101–050103 CrossRef.
- I. Singh, P. K. Bhatnagar, P. C. Mathur, I. Kaur, L. M. Bharadwaj and R. Pandey, Carbon, 2008, 46, 1141–1144 CrossRef CAS.
- E. Kymakis and G. A. J. Amaratunga, Rev. Adv. Mater. Sci., 2005, 10, 300–305 CAS.
- M. Al-Ibrahim, H. K. Roth, M. Schroedner, A. Konkin, U. Zhokhavets, G. Gobsch, P. Scharff and S. Sensfuss, Org. Electron., 2005, 6, 65–77 CrossRef CAS.
- A. Nish, J. Y. Hwang, J. Doig and R. J. Nicholas, Nat. Nanotechnol., 2007, 2, 640–646 CrossRef CAS.
- F. Y. Cheng, P. Imin, C. Maunders, G. Botton and A. Adronov, Macromolecules, 2008, 41, 2304–2308 CrossRef CAS.
- A. Nish, J. Y. Hwang, J. Doig and R. J. Nicholas, Nanotechnology, 2008, 19, 095603–095603 CrossRef.
- J. Y. Hwang, A. Nish, J. Doig, S. Douven, C. W. Chen, L. C. Chen and R. J. Nicholas, J. Am. Chem. Soc., 2008, 130, 3543–3553 CrossRef CAS.
- F. M. Chen, B. Wang, Y. Chen and L. J. Li, Nano Lett., 2007, 7, 3013–3017 CrossRef CAS.
- N. A. Rice, K. Soper, N. Z. Zhou, E. Merschrod and Y. M. Zhao, Chem. Commun., 2006, 4937–4939 RSC.
- Y. K. Kang, O. S. Lee, P. Deria, S. H. Kim, T. H. Park, D. A. Bonnell, J. G. Saven and M. J. Therien, Nano Lett., 2009, 9, 1414–1418 CrossRef CAS.
- M. Leclerc, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 2867–2873 CrossRef CAS.
- E. Lim, B. J. Jung and H. K. Shim, Macromolecules, 2003, 36, 4288–4293 CrossRef CAS.
- E. Lim, B. J. Jung, J. Lee, H. K. Shim, J. I. Lee, Y. S. Yang and L. M. Do, Macromolecules, 2005, 38, 4531–4535 CrossRef CAS.
- M. Inbasekaran, E. Woo, W. S. Wu, M. Bernius and L. Wujkowski, Synth. Met., 2000, 111, 397–401 CrossRef.
- M. Bernius, M. Inbasekaran, E. Woo, W. S. Wu and L. Wujkowski, J. Mater. Sci.: Mater. Electron., 2000, 11, 111–116 CrossRef CAS.
- M. Bernius, M. Inbasekaran, E. Woo, W. S. Wu and L. Wujkowski, Thin Solid Films, 2000, 363, 55–57 CrossRef CAS.
- R. D. McCullough, J. Polym. Sci., Part A: Polym. Chem., 1998, 10, 93–116 CAS.
- I. F. Perepichka, D. F. Perepichka, H. Meng and F. Wudl, Adv. Mater., 2005, 17, 2281–2305 CrossRef CAS.
- B. Liu, W. L. Yu, Y. H. Lai and W. Huang, Macromolecules, 2000, 33, 8945–8952 CrossRef CAS.
- M. Trznadel, A. Pron and M. Zagorska, Macromolecules, 1998, 31, 5051–5058 CrossRef CAS.
- D. Fujishima, T. Mori, T. Mizutani, T. Yamamoto and N. Kitamura, Jpn. J. Appl. Phys., 2005, 44, 546–550 CrossRef CAS.
- N. Kiriy, E. Jahne, H.-J. Adler, M. Schneider, A. Kiriy, G. Gorodyska, S. Minko, D. Jehnichen, P. Simon, A. A. Fokin and M. Stamm, Nano Lett., 2003, 3, 707–712 CrossRef CAS.
- S. Gunes, H. Neugebauer and N. S. Sariciftci, Chem. Rev., 2007, 107, 1324–1338 CrossRef.
- D. H. Zhang, K. Ryu, X. L. Liu, E. Polikarpov, J. Ly, M. E. Tompson and C. W. Zhou, Nano Lett., 2006, 6, 1880–1886 CrossRef CAS.
- Z. C. Wu, Z. H. Chen, X. Du, J. M. Logan, J. Sippel, M. Nikolou, K. Kamaras, J. R. Reynolds, D. B. Tanner, A. F. Hebard and A. G. Rinzler, Science, 2004, 305, 1273–1276 CrossRef CAS.
- J. Chen, M. A. Hamon, H. Hu, Y. S. Chen, A. M. Rao, P. C. Eklund and R. C. Haddon, Science, 1998, 282, 95–98 CrossRef CAS.
- F. Torrens, Nanotechnology, 2005, 16, 181–189.
- J. L. Bahr, E. T. Mickelson, M. J. Bronikowski, R. E. Smalley and J. M. Tour, Chem. Commun., 2001, 193–194 RSC.
- M. S. Dresselhaus, G. Dresselhaus, A. Jorio, A. G. Souza and R. Saito, Carbon, 2002, 40, 2043–2061 CrossRef CAS.
- D. A. Heller, P. W. Barone, J. P. Swanson, R. M. Mayrhofer and M. S. Strano, J. Phys. Chem. B, 2004, 108, 6905–6909 CrossRef CAS.
- S. M. Bachilo, M. S. Strano, C. Kittrell, R. H. Hauge, R. E. Smalley and R. B. Weisman, Science, 2002, 298, 2361–2366 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Full experimental details for the synthesis of monomers and polymers, as well as UV-Vis absorption results for polymers at different temperatures. See DOI: 10.1039/c0py00286k |
| ‡ This paper is part of a Polymer Chemistry issue highlighting the work of emerging investigators in the polymer chemistry field. Guest Editors: Rachel O'Reilly and Andrew Dove. |
| This journal is © The Royal Society of Chemistry 2011 |