Polymerization of an activated ester monomer based on 4-vinylsulfonic acid and its polymer analogous reaction†‡
Katja
Nilles
a and
Patrick
Theato
*ab
aJohannes Gutenberg-University Mainz, Institute of Organic Chemistry, Duesbergweg 10-14, D-55099, Mainz, Germany. E-mail: theato@uni-mainz.de; Fax: +49-6131-3924778; Tel: +49-6131-3926256
bSchool of Chemical and Biological Engineering, WCU program of Chemical Convergence for Energy & Environment (C2E2), College of Engineering, Seoul National University, 151-744, Seoul, Korea
First published on 15th October 2010
Abstract
Homopolymers containing sulfonic ester side groups were synthesized by reversible addition–fragmentation chain transfer (RAFT) polymerization utilizing benzyl dithiobenzoate, cumyl dithiobenzoate, and 4-cyano-4-((thiobenzoyl)sulfanyl)pentanoic acid as chain transfer agents. Likewise diblock copolymers containing poly(styrene), poly(octylstyrene) and poly(pentafluorostyrene) as the second block were synthesized. Additionally, nitroxide mediated polymerization (NMP) was investigated for the synthesis of a homopolymer as well as for a diblock copolymer. Furthermore, the post-polymerization functionalization with various amines to yield the respective sulfonamides was conducted. The conversion was analyzed by 1H NMR spectroscopy, 19F NMR spectroscopy and FT-IR spectroscopy and in many cases a very high conversion (>96%) was observed. In addition the reaction kinetics of the post-polymerization functionalization of poly(pentafluorophenyl 4-vinylbenzene sulfonate) and the corresponding carboxyl ester poly(pentafluorophenyl 4-vinylbenzoate) were compared by analysis of the reactions by time-resolved 19F NMR spectroscopy. It was found that poly(pentafluorophenyl 4-vinylbenzoate) showed a higher stability towards hydrolysis and a significantly higher reactivity, resulting in complete conversions with different amines.
Introduction
Sulfonic acid esters find application in various areas, e.g. as precursors for pharmaceutically interesting sulfonamides.1,2 Low molecular weight compounds containing sulfonamides as structural motifs are known for their antimicrobial and biological activity.3–5Sulfonamides are usually synthesized by reacting sulfonyl chlorides with amines. Difficulties in the preparation using highly reactive sulfonyl chlorides were recently circumvented through the use of the respective activated esters.1,6–9 These activated esters replaced sulfonyl chlorides because of their easier handling, longer storability and higher tolerance towards reaction conditions.10 Caddick and coworkers determined the reactivity of low molecular weight pentafluorophenyl and trichlorophenyl sulfonate esters being converted with a wide range of amines, thereby developing optimal reaction conditions e.g. by utilizing chloride salts, which accelerated the rate of aminolysis.6–9,11Furthermore, sulfonic acid esters have been used as protective groups that allow for the polymerization of the respective sulfonic acid. In a post-polymerization reaction those esters can be hydrolyzed thermally or under basic conditions.12–15
However, to the best of our knowledge, polymeric activated esters of sulfonic acids have not been explored in the preparation of functional polymers. Accordingly, we herein report on the synthesis of polymers that utilize activated esters of sulfonic acid as reactive groups that can be used to obtain the respective polymeric sulfonamides. To show the applicability of activated esters of sulfonic acid in polymer synthesis we will investigate the preparation of homo- and block copolymers by reversible addition–fragmentation chain transfer (RAFT) polymerization as well as the polymer analogous reaction of two different sulfonic acid esters, pentafluorophenyl 4-vinylbenzene sulfonate and phenol 4-vinylbenzene sulfonate. Finally, we will compare these two sulfonic acid esters with pentafluorophenyl 4-vinylbenzoate in terms of reactivity.
Results and discussion
In the literature various sulfonate containing polymers are known, however, only few examples of monomers carrying sulfonates in the side chain, e.g.sodium p-styrene sulfonate16–18 and neopentyl styrene sulfonate,12 have been polymerized by radical polymerization techniques. Therefore, it was desirable to explore sulfonic acid ester based monomers and we hence synthesized two sulfonic acid esters, pentafluorophenyl 4-vinylbenzene sulfonate (PFPVS) and phenyl 4-vinylbenzene sulfonate (PVS), which are expected to react with amines yielding the respective sulfonamides (see Scheme 1). For the synthesis of the monomers, commercially available sodium p-styrene sulfonate was first converted into the sulfonyl chloride,19 which was in a next step used to react with pentafluorophenol or phenol to yield the respective sulfonic ester.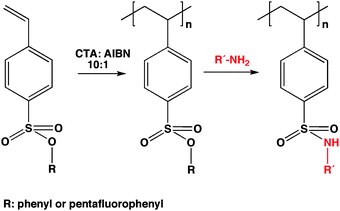 | ||
| Scheme 1 General scheme of the polymerization and polymer analogous conversion of the polymeric sulfonate esters. | ||
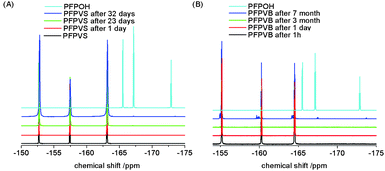
In order to develop suitable conditions for polymerization and post-polymerization functionalization, i.e.polymer analogous reactions, the hydrolysis of the activated ester monomers was analyzed first (see Fig. 1). In this respect the hydrolysis of pentafluorophenyl 4-vinylbenzene sulfonate (PFPVS) was compared to the already established activated ester monomer pentafluorophenyl 4-vinylbenzoate (PFPVB).20,21 The respective monomer was dissolved in dioxane and then water (15 vol%) was added. Time dependent 19F NMR spectra were recorded at room temperature. The time at which the first detectable signals other than the three signals of the pentafluorophenyl ester were detected was assigned as the onset time of hydrolysis. For PFPVS peak broadening as well as peaks around −167 ppm could be observed after 32 days. In contrast, the first additional peaks for the hydrolysis of PFPVB were observed after more than 3 months. As a comparison the 19F NMR spectrum of free pentafluorophenol (PFPOH) was measured, however, it has to be noticed that the signals of free pentafluorophenol are shifted dramatically when compared to (i) the two pentafluorophenyl esters and (ii) the additional signals occurring during hydrolysis, which is likely due to the difference in chemical environment as 19F NMR spectroscopy is extremely sensitive to changes in the chemical environment or to the presence of other compounds. Nevertheless, it can be stated that the PFPVS is stable for up to 32 days in an aqueous environment at room temperature. Compared to the PFPVB, which is stable for up to three month, PFPVS appears to be more sensitive in respect to hydrolysis, however, a stability of about 1 month in aqueous solution at neutral pH makes this reactive group still very attractive for further polymer functionalization chemistries.
In continuation of our initiatives to control architecture and functionality of polymers by polymer analogous reactions of well-defined polymers,21–23 it was very interesting to study the controlled polymerization of PFPVS. In a first attempt, the homopolymerization of both sulfonic acid ester based monomers, PFPVS and PVS, was conducted under RAFT polymerization conditions. The polymerization was conducted in solution using benzyl dithiobenzoate (CTA1), cumyl dithiobenzoate (CTA2) or 4-cyano-4-(thiobenzoylthio) valeric acid (CTA3) as the chain transfer agents and AIBN as the radical initiator for around 18 h. The obtained polymers are listed in Table 1, with reported yields after precipitation and drying in vacuum. In all cases polymers with well-defined molecular weight ranging between 4200 and 28200 g mol−1 while maintaining in many cases a narrow molecular weight distribution (Mw/Mn = 1.07–1.41) were obtained. However, the yields differed significantly. The RAFT polymerization of PVS or PFPVS utilizing CTA1 and CTA2 as chain transfer agents resulted in acceptable yields of up to 55%. However, the use of CTA3 for the RAFT polymerization of PFPVS resulted only in a very moderate yield of 6% as can be seen for polymer P1d in Table 1.
| # | Polymer | CTAb | Yieldc | M n,GPC | M w,GPC | PDI | RatioNMR |
|---|---|---|---|---|---|---|---|
| a GPC measured in THF, all others were measured in DMF. b CTA1: benzyl dithiobenzoate; CTA2: cumyl dithiobenzoate; CTA3: 4-cyano-4-(thiobenzoylthio) valeric acid; and CTA4: 4-cyano-4-dodecylsulfanylthiocarbonylsulfanyl-4-methyl-butyric acid. c Yield after precipitation and drying. | |||||||
| P1a | PolyPFPVS | CTA1 | 51% | 8700 | 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700 700 |
1.33 | — |
| P1b | PolyPFPVS | CTA1 | 55% | 28200 |
36100 |
1.28 | — |
| P1c | PolyPFPVS | CTA2 | 32% | 7600 | 8500 | 1.11 | — |
| P1d | PolyPFPVS | CTA3 | 6% | 14000 |
17300 |
1.23 | — |
| P1e | PolyPFPVS | MAMA | 51% | 25700 |
33600 |
1.31 | — |
| P2a | PolyPVS | CTA1 | 25% | 4200 | 5400 | 1.25 | — |
| P2b | PolyPVS | CTA1 | 20% | 24000 |
33800 |
1.41 | — |
| P2c | PolyPVS | CTA2 | 34% | 5200 | 5500 | 1.07 | — |
| P2d a | PolyPVS | CTA2 | 51% | 9600 | 12200 |
1.27 | — |
| P3a | PolyPFPVB | CTA2 | 63% | 17400 |
22700 |
1.31 | — |
| P4 | Poly(styrene) | CTA3 | 24% | 12700 |
15100 |
1.19 | — |
| P6 | PolyPFS | CTA4 | 56% | 1500 | 1600 | 1.10 | — |
| P8a | PolyOS | MAMA | 40% | 23500 |
34300 |
1.46 | — |
| P5 | Poly(styrene)-b-polyPFPVS | P4 | 18% | 21100 |
28200 |
1.34 | 1:0.11 |
| P7 | PolyPFS-b-polyPFPVS | P6 | 6% | 17800 |
21500 |
1.20 | 1:3.03 |
| P9 | PolyOS-b-polyPFPVS | P8 | 13% | 69200 |
101500 |
1.47 | 1:0.35 |
| P10 | PolyPFPVS-b-poly(styrene) | P1a | 6% | 14000 |
16100 |
Bimodal | 1:2.17 |
| 2800 | 3000 |
These results motivated for a detailed kinetic analysis of the RAFT polymerization of PFPVS (M1) using CTA1, which resulted in the best yields with acceptable Mw/Mn. The results of the kinetic investigation are summarized in Fig. 2. From aliquots of a polymerization solution in THF containing monomer, chain transfer agent (CTA1) and AIBN as initiator, which had been taken at regular time intervals, the conversion and molecular weight were determined. To evaluate the polymerization kinetics ln(M0/M) as a function of time and the molecular weight distribution Mw/Mn as a function of monomer conversion were plotted and are shown in Fig. 2. These first-order kinetic plots showed a linear behavior up to 8 h corresponding to a conversion of about 50%. Even though the molecular weight distribution is with values around Mw/Mn = 1.4 a little broader than usually, all in all the polymerization kinetics of the RAFT polymerization of PFPVS show a controlled behavior. If higher conversions for the polymerization were targeted, then the kinetics deviated from the linear behavior indicating that the controlled behavior was not perfect. The RAFT polymerization of PFPVS was investigated using three chain transfer agents CTA1, CTA2 and CTA3, however, only the use of CTA1 as the chain transfer agent resulted in good yields. It can be concluded that a controlled behavior for the RAFT polymerization of PFPVS is indicated if CTA1 is used as a chain transfer agent and if conversions are kept below 50%.
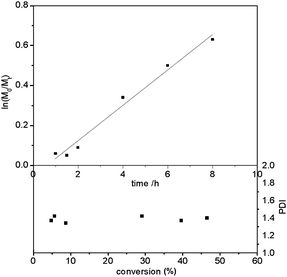 | ||
| Fig. 2 Kinetic studies for the RAFT polymerization of PFPVS using chain transfer agent CTA1 in THF at 80 °C. ln(M0/Mt) versus time and evolution Mw/Mn values with conversion. | ||
In comparison, the RAFT polymerization of PVS showed a better control when using cumyl dithiobenzoate (CTA2) as the chain transfer agent. The molecular weight distribution was very narrow (Mn/Mw = 1.07) for polymerizations using CTA2, while a comparable RAFT polymerization using benzyl dithiobenzoate (CTA1) resulted in a not as narrow but still acceptable molecular weight distribution (Mn/Mw = 1.25). Furthermore, nitroxide-mediated polymerization (NMP) was briefly investigated to synthesize a homopolymer of PFPVS by utilization of BlocBuilder™ (MAMA). The synthesized homopolymer had a molecular weight of Mn = 25700 g mol−1 with a narrow molecular weight distribution of Mn/Mw = 1.31 and could be prepared with acceptable yields (51%).
Furthermore, the solubility of the obtained homopolymers was investigated. They showed an excellent solubility in most organic solvents as for example THF, chloroform, dichloromethane, acetone and DMF. For precipitation methanol, hexane and dioxane could be used. To determine the thermal stability of the polymeric sulfonate esters, thermogravimetric analysis (TGA) was performed. The TGA curves of the three polymers polyPFPVS, polyPVS and polyPFPVB are shown in Fig. 3. All three polymers are very stable up to 250 °C, indicating a broad temperature window for potential polymer analogous reactions. While polyPVS and polyPFPVB began to decompose at temperatures above 350 °C, polyPFPVS seemed to be less temperature stable as a first decomposition could be observed around 300 °C. However, for all three polymers, no defined decomposition reaction could be assigned.
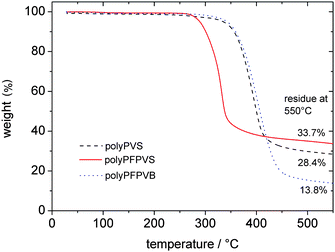 | ||
| Fig. 3 TGA of polyPFPVS, polyPVS and polyPFPVB. | ||
Next, to take advantage of the described control of the polymerizations of PFPVS, the synthesis of different block copolymers of PFPVS using RAFT and nitroxide mediated polymerization (NMP) techniques was briefly investigated. The results are summarized in the second half of Table 1. At first, the synthesis of polyPFPVS-b-poly(styrene) was performed utilizing CTA1 as the chain transfer agent for the polymerization of PFPVS as the first block (P1a), followed by reinitiating the polymerization of styrene as the second block, yielding the diblock copolymer P10. However, a bimodal block copolymer was obtained with molecular weights of Mn = 14000 g mol−1 and Mn = 2800 g mol−1 in a very low yield (6%). A block ratio of 1:2.17 was calculated by 1H NMR measurement. After this not successful experimental attempt, polymerizations of block copolymers with PFPVS as the second block were investigated. In the attempt to synthesize poly(styrene)-b-polyPFPVS by RAFT polymerization of PFPVS utilizing poly(styrene) (P4) as the macro-CTA only a low conversion of the second monomer PFPVS could be achieved and a block ratio of 1:0.11 was calculated from the 1H NMR spectrum. Calculation of the block ratio from the molecular weight resulted in slightly higher ratios (1:0.2 for Mn and 1:0.26 for Mw), likely due to the fact that the molecular weights calculated by GPC are apparent molecular weights. Only the RAFT polymerization of poly(pentafluorostyrene)-b-polyPFPVS resulted in a good incorporation of PFPVS as the second block using poly(pentafluorostyrene) (P6) as the macro-CTA reaching a block ratio of 1:3.03 (determined by 19F NMR), with narrow molecular weight distributions of Mw/Mn = 1.20. However, the yield was relatively low (6%) due to difficulties in the precipitation.
Similar results were obtained for the synthesis of poly(octylstyrene)-b-polyPFPVS by NMP. A block copolymer could be obtained with a molecular weight of 69200 g mol−1 by polymerization of PFPVS using poly(octylstyrene) (P8) (Mn = 23500 g mol−1) as the macroinitiator. However, the molecular weight distribution was a bit broader (Mn/Mw = 1.47), which resulted from the broader molecular weight distribution (Mn/Mw = 1.46) of the macroinitiator poly(octylstyrene). Nevertheless, a better block ratio of 1:0.35 (determined by 1H NMR) could be achieved. It thus can be concluded that it is in principle possible to synthesize block copolymers using PFPVS as second monomer. The reinitiation was possible in the synthesis of block copolymers, but the polymerization conditions need to be optimized in order to reach higher conversion of the second monomer, which is beyond the scope of the present study.
In the end, the polymer analogous reaction of polyPFPVS as well as polyPVS with different amines was studied. Table 2 outlines the results of those reactions with different amines as well as KOH. In contrast to previous investigations of the polymer analogous reactions of polyPFPVB with amines,20 not all reactions of the polyPFPVS with amines resulted in quantitative conversions. This can be demonstrated by the superior analytical advantage of 19F NMR over 1H NMR for these kinds of investigations with the data given in the experimental section. While in many cases a complete conversion can be calculated from the 1H NMR spectra, the analysis of the 19F NMR spectra revealed still unreacted pentafluorophenyl activated ester moieties, which was further confirmed by IR spectroscopy. For the reactions with potassium hydroxide and N,N-dimethylethylenediamine quantitative conversion could be determined, while for the reactions with isopropylamine, hexylamine, cyclopropylamine, morpholine, pyrrolidine and aniline conversions over 96% (up to 99%) have been measured. As long as a quantitative conversion of the reactive groups is not required, the reaction still represents an outstanding possibility to prepare different classes of functional polymers. Even though, the reaction of polyPFPVS with the aromatic amine aniline resulted in 96% conversion, the reaction with the secondary amine diethylamine resulted only in 63%, which indicates a clear differentiation between primary and secondary amines. It is further noteworthy that the reactions of the respective amines with polyPVS showed no conversion, not even when conducted at 60 °C. This demonstrates the advantageous use of activated esters also for polymeric sulfonates.
| Polymer | Reactive polymer | Amine | Conversionb |
|---|---|---|---|
| a The reaction was performed at 60 °C. b The conversion was determined by 1H NMR and IR-spectroscopy. | |||
| PA1 | PolyPFPVS | KOH | 100% |
| PA2 | PolyPFPVS | Isopropylamine | >99% |
| PA3 | PolyPFPVS | Dimethylethylenediamine | 100% |
| PA4 | PolyPFPVS | Hexylamine | >96% |
| PA5 | PolyPFPVS | Cyclopropylamine | >99% |
| PA6 | PolyPFPVS | Morpholine | >96% |
| PA7 | PolyPFPVS | Pyrrolidine | >96% |
| PA8 | PolyPFPVS | Diethylamine | 63% |
| PA9 a | PolyPFPVS | Aniline | >96% |
| PA10 a | PolyPVS | Isopropylamine | 0% |
| PA11 a | PolyPVS | Diisopropylamine | 0% |
| PA12 a | PolyPVS | Dimethylethylenediamine | 0% |
| PA13 | PolyPFPVB | Dimethylethylenediamine | 100% |
As an example, the analysis of the conversion of the reaction of polyPFPVS with N,N-dimethylethylenediamine yielding poly((N,N-dimethylaminoethyl) 4-vinylbenzene sulfonamide) (PA3) will be described in the following. In the 1H NMR spectrum, measured after the aminolysis, the peaks appearing at 2.96 ppm and 2.36 ppm for the methylene-groups and at 2.13 ppm for the methyl-groups could be assigned to poly(N,N-dimethylaminoethyl 4-vinylbenzene sulfonamide). In the 19F NMR spectrum no fluorine signals could be observed due to the occurred cleavage of pentafluorophenol. The FT-IR spectrum of polyPFPVS showed the typical sulfonate bands at 1390 cm−1 and 1182 cm−1, as well as the C–F band at 1196 cm−1. After the successful conversion new bands appeared, most importantly the N–CH3 band at 2805 cm−1, whereas the C–F band at 1196 cm−1 vanished.
Next, the reactions of the polymeric sulfonate ester polyPFPVS with N,N-dimethylethylenediamine was compared to the reaction of the polymeric carboxylic ester polyPFPVB with N,N-dimethylethylenediamine. Both conversions have been conducted at room temperature in solution. The conversion was verified by 19F NMR, 1H NMR and IR spectroscopy. 19F NMR spectroscopy was used exemplary to follow the kinetics of both conversions and the results are shown in Fig. 4. The conversion was monitored by the decrease of the signal intensities of the three signals at −152 ppm, −156 ppm and −162 ppm for polyPFPVS and −154 ppm, −158 ppm and −163 ppm for polyPFPVB, which are in both cases indicative for the pentafluorophenyl ester. The reaction of the polyPFPVS seemed to be considerably slower compared to the reaction of the corresponding carboxylic ester (see Fig. 4). Fig. 4a shows time resolved 19F NMR spectra of the reaction of polyPFPVS and begins with the unconverted species (100% polyPFPVS). After 58 minutes more than half of the activated ester (49% remaining) is converted to the amide and after 99 minutes full conversion is achieved. Contrary, the reaction of polyPFPVB (Fig. 4b) showed already a conversion of 53% after 7 minutes and after 47 minutes a quantitative conversion was determined.
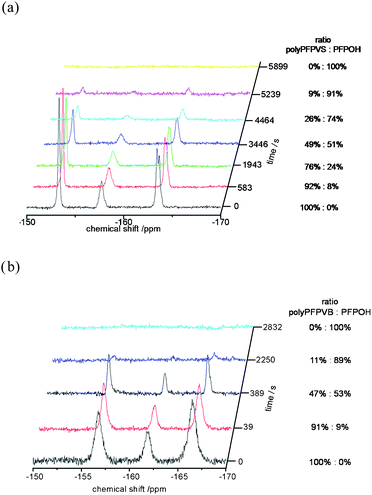 | ||
| Fig. 4 19F NMR kinetic studies of the conversion of the active ester with N,N-dimethylethylenediamine (a) conversion of polyPFPVS and (b) conversion of polyPFPVB. | ||
Conclusions
Poly(pentafluorophenyl 4-vinylbenzene sulfonate) (polyPFPVS) synthesized viaRAFT polymerization or NMP represents a new class of sulfonate containing polymers that can be used to prepare polymeric sulfonamidesvia a polymer analogous reaction with amines. To obtain full conversion an excess amount of amine is necessary. PolyPVS instead does not react under the same reaction conditions with amines. When comparing the sulfonate activated ester with the carboxylic acid activated ester it was found that the post-polymerization functionalization of the carboxylic acid ester is significantly faster than the sulfonate analogue. Nevertheless, in both cases full conversion at room temperature can be achieved.Experimental section
Materials
All chemicals were commercially available and used as received unless otherwise stated. 2,2′-Azoisobutyronitrile (AIBN) was recrystallized from diethyl ether. All solvents were purified by common procedures. Styrene and 2,3,4,5,6-pentafluorostyrene were used freshly distilled. 4-Octylstyrene was synthesized according to a previously published procedure.24MAMA (BlocBuilder™) was kindly provided by Arkema (France). Benzyl dithiobenzoate (CTA1),25–27cumyl dithiobenzoate (CTA2),25–274-cyano-4-(thiobenzoylthio) valeric acid (CTA3),28 and 4-cyano-4-dodecylsulfanylthiocarbonylsulfanyl-4-methyl-butyric acid (CTA4)29 were synthesized as described in the literature.Instrumentation
1H and 13C NMR spectra were obtained on a Bruker AC 300 MHz FT-NMR spectrometer, and 19F NMR spectra on a Bruker AC 376 MHz FT-NMR spectrometer. Gel permeation chromatography (GPC) was used to determine molecular weights and polydispersity indices (PDI), Mw/Mn, of polymeric samples with respect to polystyrene standards. Therefore, a GPC set-up was used consisting of the following components: a Jasco PU-1580 pump, a Jasco AS-1555 autosampler, MZ-Gel-SDplus columns (102, 104 and 106 Å2), a Jasco RI-1530 refractive index detector, and a Jasco UV-1575 UV/vis detector. Infrared (IR) spectra were measured on a Bruker Vector 22 FT-IR spectrometer with ATR unit. Thermo gravimetrical analysis was performed using a Perkin Elmer Pyris 6 TGA in nitrogen (10 mg pure polymer in aluminium pan).p-Styrenesulfonyl chloride
A mixture of 50 mL (0.69 mol) thionyl chloride and 0.3 g (1,4 mmol) 2,6-di-tert-butyl-4-methylphenol in 60 mL (0.77 mol) anhydrous N,N-dimethylformamide (DMF) was stirred in an ice bath. 20 g of (86 mmol) 4-vinylbenzenesulfonic acid sodium salt were added in small portions and the solution was stirred for 3 hours at 0 °C to afford a homogeneous solution. After standing for 24 h in a refrigerator, the reaction mixture was slowly poured into 300 mL ice water and extracted twice with benzene (each 200 mL). The extract was washed twice with water (each 200 mL), dried over anhydrous sodium sulfate, and concentrated on a rotary evaporator below 40 °C. A pale yellow liquid was obtained in a quantitative yield, which was used immediately without further characterization.Pentafluorophenyl 4-vinylbenzene sulfonate (M1)
To 6.9 g (37 mmol) pentafluorophenol and 5.2 mL (3.8 g; 38 mmol) triethylamine in 30 mL dichlormethane, 7 g (35 mmol) of p-styrenesulfonyl chloride were added slowly at 0 °C under nitrogen. Afterwards the solution was stirred for 3 hours at 0 °C, followed by stirring for 24 hours at room temperature. Afterwards the reaction mixture was washed twice with water and the organic phase was dried over anhydrous MgSO4. After evaporation of the solvent, the residue was purified by column chromatography using DCM:hexane (1:1). The product was obtained as pale yellow crystals (yield: 8.53 g; 71%).
1H NMR (300 MHz, CDCl3): δ (ppm) = 7.92 (d, 2H, 3J = 8.5 Hz) (benzene sulfonate, ortho); 7.61 (d, 2H, 3J = 8.5 Hz) (benzene sulfonate, meta); 6.79 (dd, 1H, 3Jtrans = 17.7 Hz, 3Jcis = 11.0 Hz) (vinyl, –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) ); 5.96 (d, 1H, 3J = 17.7 Hz) and 5.53 (d, 1H, 3J = 10.7 Hz) (vinyl, CH2); 13C NMR (75 MHz, CDCl3): δ (ppm) = 144.44 (C–S); 143.09, 141.92, 140.68, 139.70, 138.58, 136.18 (pentafluorophenyl, ortho, meta and para); 134.91 (CH from CHCH2); 133.25 (CO aromatic); 128.90 (CH benzene sulfonate, meta); 127.03 (CH benzene sulfonate, ortho); 119.09 (CH2); 19F NMR (376 MHz, CDCl3): δ (ppm) = −150.90 (d, 2F, 3J = 17.1 Hz) (pentafluorophenyl, ortho); −155.77 (t, 1F, 3J = 18.8 and 21.9 Hz) (pentafluorophenyl, para); −161.46 (dd, 2F, 3J = 17.1 and 22.6 Hz) (pentafluorophenyl, meta). Elemental analysis: (C14H7F5O3S) (350.26): calcd C 48.01, H 2.01, F 27.12, O 13.70, S 9.15; found C 47.92, H 1.94, S 8.89%; mp: 52 °C.
); 5.96 (d, 1H, 3J = 17.7 Hz) and 5.53 (d, 1H, 3J = 10.7 Hz) (vinyl, CH2); 13C NMR (75 MHz, CDCl3): δ (ppm) = 144.44 (C–S); 143.09, 141.92, 140.68, 139.70, 138.58, 136.18 (pentafluorophenyl, ortho, meta and para); 134.91 (CH from CHCH2); 133.25 (CO aromatic); 128.90 (CH benzene sulfonate, meta); 127.03 (CH benzene sulfonate, ortho); 119.09 (CH2); 19F NMR (376 MHz, CDCl3): δ (ppm) = −150.90 (d, 2F, 3J = 17.1 Hz) (pentafluorophenyl, ortho); −155.77 (t, 1F, 3J = 18.8 and 21.9 Hz) (pentafluorophenyl, para); −161.46 (dd, 2F, 3J = 17.1 and 22.6 Hz) (pentafluorophenyl, meta). Elemental analysis: (C14H7F5O3S) (350.26): calcd C 48.01, H 2.01, F 27.12, O 13.70, S 9.15; found C 47.92, H 1.94, S 8.89%; mp: 52 °C.
Phenyl 4-vinylbenzene sulfonate (M2)
Phenyl 4-vinylbenzene sulfonate was synthesized in analogy to the fluorinated monomer by using phenol instead of pentafluorophenol (yield: 79%).
1H NMR (300 MHz, CDCl3): δ (ppm) = 7.75 (d, 2H, 3J = 8.5 Hz) (benzene sulfonate, ortho); 7.50 (d, 2H, 3J = 8.5 Hz) (benzene sulfonate, meta); 7.33–7.16 (m, 3H) (phenyl, meta and para); 6.97 (d, 2H, 3J = 7.9 Hz) (phenyl, ortho); 6.73 (dd, 1H, 3Jtrans = 17.6 Hz, 3Jcis = 10.9 Hz) (vinyl, –CH); 5.90 (d, 1H, 3J = 17.3 Hz) and 5.46 (d, 1H, 3J = 10.9 Hz) (vinyl, CH2); 13C NMR (75 MHz, CDCl3): δ (ppm) = 149.59 (C–O); 143.28 (benzene sulfonate, ipso, C–S); 135.09 (benzene sulfonate, para, next to CHCH2); 134.01 (CH from CHCH2); 129.67 (phenyl, meta); 128.86 (benzene sulfonate, ortho); 127.18 (benzene sulfonate, meta); 126.67 (phenyl, para); 122.37 (phenyl, ortho); 118.37 (CH2). Elemental analysis: (C14H12O3S) (260.31): calcd C 64.60, H 4.65, O 18.44, S 12.32; found C 64.34, H 4.72, S 12.24%; mp: 33.1 °C.
Pentafluorophenyl 4-vinylbenzoate (M3)
Pentafluorophenyl 4-vinylbenzoate was synthesized as published previously.20 In the first step 4-vinylbenzoyl chloride (4VBC) was prepared by reacting 4-vinylbenzoic acid with oxalyl chloride and in a second step 4VBC was esterificated with 2,3,4,5,6-pentafluorophenol. Pentafluorophenyl 4-vinylbenzoate was then purified by column chromatography using toluene: n-hexane (1:1) as an eluent.
1H NMR (300 MHz, CDCl3): δ (ppm) = 8.15 (d, 2H, 3J = 8.5 Hz) (benzoate, para); 7.55 (d, 2H, 3J = 8.5 Hz) (benzoate, meta); 6.79 (dd, 1H, 3Jtrans = 17.7 Hz, 3Jcis = 11.0 Hz) (vinyl, CHCH2); 5.94 (d, 1H, 3J = 17.3 Hz) and 5.47 (d, 1H, 3J = 10.7 Hz) (vinyl, CH2CH–). 13C NMR (75 MHz, CDCl3): δ (ppm) = 162.5 (COO); 143.7 (benzoate, para); 143.1, 141.2, 139.7, 137.8, 136.3 (pentafluorophenyl, ortho, meta and para); 135.6 (vinyl, –CHCH2); 131.1 (benzoate, ortho); 126.5 (benzoate, meta and ipso); 125.8 (pentafluorophenyl, ipso); 117.7 (vinyl, CH2CH–). 19F NMR (376 MHz, CDCl3): δ (ppm) = −152.89 (d, 2F, 3J = 18.4 Hz) (pentafluorophenyl, ortho); −158.48 (t, 1F, 3J = 23.0 and 20.0 Hz) (pentafluorophenyl, para); −162.84 (dd, 2F, 3J = 20.7 and 22.9 Hz) (pentafluorophenyl, meta). Elemental analysis: (C15H7F5O2) (314.21): calcd C 57.34, H 2.25, F 30.23, O 10.18; found C 57.25, H 2.19%; mp: 39 °C.
Hydrolysis
Hydrolysis experiments have been conducted using PFPVS and PFPVB by dissolving 20 mg of the respective monomer in 0.6 mL deuterated dioxane. After the addition of 0.1 mL of H2O 19F NMR spectra were recorded in time intervals until the first signs of hydrolysis could be detected. The NMR tubes were stored in between the experiments at room temperature.Polymerization
(A) For the RAFT polymerization the respective monomer was dissolved in 2 mL freshly distilled dry solvent in a Schlenk flask. AIBN and the respective chain transfer agent (CTA) (molar ratio of AIBN/CTA 1:10) were added and after degassing by three freeze–pump–thaw cycles, the flask was immersed in a preheated oil bath at 80 °C for a given time. After cooling to room temperature, the polymer was isolated by precipitation and was dried in vacuum.
(B) For NMP a Schlenk flask was filled with monomer (5.7 mmol) and MAMA (8 × 10−2 mmol) in 3 mL freshly distilled THF, degassed by three freeze–pump–thaw cycles. Afterwards the flask was immersed in a preheated oil bath at 120 °C and the polymerization was conducted for 24 h (homopolymers) or 72 h (block copolymers), respectively. After cooling to room temperature, the polymer was isolated by precipitation and was dried in vacuum. If not otherwise mentioned RAFT polymerization was used for the preparation of the polymer.
Poly(pentafluorophenyl 4-vinylbenzene sulfonate) (P1)
1H NMR (300 MHz, DMSO): δ (ppm) = 7.90–7.43 (br, 2H) (benzene sulfonate, ortho); 7.43–6.58 (br, 2H) (benzene sulfonate, meta); 2.22–1.17 (br, 3H) (backbone); 19F NMR (377 MHz, DMSO): δ (ppm) = −152.16 to −153.38 (br, 2F) (pentafluorophenyl sulfonate, ortho); −155.73 to −156.85 (br, 1F) (pentafluorophenyl sulfonate, para); −161.85 to −162.88 (br, 2F) (pentafluorophenyl sulfonate, meta). FT-IR (cm−1): 2933 (aryl); 1390 and 1182 (sulfonate); 1196 (C–F). GPC (in DMF): Mn = 8700 g mol−1, Mw = 12700 g mol−1, Mw/Mn = 1.33 (P1a); Mn = 28200 g mol−1, Mw = 36100 g mol−1, Mw/Mn = 1.28 (P1b); Mn = 7600 g mol−1, Mw = 8500 g mol−1, Mw/Mn = 1.11 (P1c); Mn = 14000 g mol−1, Mw = 17300 g mol−1, Mw/Mn = 1.23 (P1d); Mn = 25700 g mol−1, Mw = 33600 g mol−1, Mw/Mn = 1.31 (P1e). The polymer (P1e) was synthesized vianitroxide mediated polymerization using MAMA as alkoxyamine, all other polymers were prepared viaRAFT polymerization. Tg: 91.9 °C. TGA: 1. step from 310.89 °C; residue at 550 °C: 33.7%
Poly(phenyl 4-vinylbenzene sulfonate) (P2)
1H NMR (300 MHz, CDCl3): δ (ppm) = 8.04–7.43 (br, 2H) (benzene sulfonate, ortho); 7.43–7.10 (br, 3H) (phenyl, meta and para); 7.10–6.82 (br, 2H) (phenyl, ortho); 6.82–6.39 (br, 2H) (benzene sulfonate, meta); 2.50–1.01 (br, 3H) (backbone); 13C NMR (75 MHz, CDCl3): δ (ppm) = 149.17 (CO); 134.18 (benzene sulfonate, para, next to CHCH2); 129.73 (benzene sulfonate, ipso, C–S); 128.78 (phenyl, meta); 128.19 (benzene sulfonate, ortho); 127.39 (benzene sulfonate, meta); 122.10 (phenyl, para and ortho), 40.89 (CH2, backbone); 29.69 (CH, backbone). FT-IR (cm−1): 2925 (aryl); 1594 and 1489 (aromatic ring); 1370 and 1171 (sulfonate). GPC (DMF): Mn = 4200 g mol−1, Mw = 5400 g mol−1, Mw/Mn = 1.25 (P2a); Mn = 24000 g mol−1, Mw = 33800 g mol−1, Mw/Mn = 1.41 (P2b); Mn = 5200 g mol−1, Mw = 5500 g mol−1, Mw/Mn = 1.07 (P2c); GPC (THF): Mn = 9600 g mol−1, Mw = 12200 g mol−1, Mw/Mn = 1.27 (P2d). Tg: 92.4 °C. TGA: 1. step from 361.3 °C; residue at 550 °C: 28.4%
Poly(pentafluorophenyl 4-vinylbenzoate) (P3)
1H NMR (300 MHz, CDCl3): δ (ppm) = 8.03–7.57 (br, 2H) (benzoate, ortho); 6.96–6.35 (br, 2H) (benzoate, meta); 2.26–1.12 (br, 3H) (backbone). 13C NMR (75 MHz, THF-d8): 162.65(CO); 152.82 (C aromatic next to the backbone); 144.01, 142.16, 140.58, 138.85, 137.26 (C aromatic pentafluorophenyl); 131.51 (CH benzoate, ortho); 129.22 (CH benzoate, meta and C next to the ester); 127.61 (C–O, pentafluorophenyl); 125.86 (CH backbone); 42.12 (CH2 backbone). 19F NMR (376 MHz, CDCl3): δ (ppm) = −152.91 to −154.04 (br, 2F) (pentafluorophenyl, ortho); −157.59 to −158.51 (br, 1F) (pentafluorophenyl, para); −162.41 to −163.51 (br, 2F) (pentafluorophenyl, meta). FT-IR (cm−1): 2932 (C–H); 1759 (CO); 1608 (aromatic ring); 1238 (C–F). GPC (THF): Mn = 17400 g mol−1, Mw = 22700 g mol−1, Mw/Mn = 1.31 (P3), yield: 63%. For P3 cumyl dithiobenzoate (CTA2) was used as RAFT agent. Tg: 99.4 °C. TGA: 1. step from 363.1 °C; residue at 550 °C: 13.8%
Poly(styrene) (P4)
1H NMR (300 MHz, CDCl3): δ (ppm) = 7.58–6.91 (br, 3H) (styrene, ortho and para); 6.91–6.27 (br, 2H) (styrene, meta); 2.55–1.13 (br, 3H) (backbone). GPC (DMF): Mn = 12700 g mol−1, Mw = 15100 g mol−1, Mw/Mn = 1.2, yield: 24%.
Poly(styrene)-b-poly(pentafluorophenyl 4-vinylbenzene sulfonate)(P5)
1H NMR (300 MHz, DMSO): δ (ppm) = 7.90–7.50 (br, 2H) (benzene sulfonate, ortho); 7.33–6.24 (br, 7H) (styrene, ortho, meta and para; benzene sulfonate, meta); 2.22–0.96 (br, 6H) (backbone). Ratio of styrene to pentafluorophenyl sulfonate was 1:0.11 from 1H NMR. 19F NMR (377 MHz, DMSO): δ (ppm) = −152.98 to −154.12 (br, 2F) (pentafluorophenyl sulfonate, ortho); −157.31 to −158.34 (br, 1F) (pentafluorophenyl sulfonate, para); −163.18 to −164.27 (br, 2F) (pentafluorophenyl sulfonate, meta). GPC (DMF): Mn = 21000 g mol−1, Mw = 28200 g mol−1, Mw/Mn = 1.34.
Poly(pentafluorostyrene)(P6)
1H NMR (300 MHz, CDCl3): δ (ppm) = 3.81–3.65 (br, 2H) (S–CH2–CH); 3.35–3.21 (br, 2H) (S–CH2–CH2); 3.00–2.86 (br, 1H) (CH–ar); 2.86–2.61 (br, 2H) (CH2–COOH); 2.61–2.20 (br, 1H) (CH, backbone); 2.20–1.72 (br, 2H) (CH2, backbone); 1.72–1.53 (br, 2H) (CH2–CH2–COOH); 1.41–1.08 (br, 20) (CH2, alkyl); 0.91–0.79 (br, 3H) (CH3). 19F NMR (376 MHz, CDCl3): δ (ppm) = −140.41 to −144.75 (br, 2F) (pentafluorostyrene, ortho); −152.73 to −154.74(br, 1F) (pentafluorostyrene, para); −159.58 to −162.33 (br, 2F) (pentafluorostyrene, meta). GPC (DMF): Mn = 1500 g mol−1, Mw = 1600 g mol−1, Mw/Mn = 1.1, yield: 57%.Poly(pentafluorostyrene)-b-poly(pentafluorophenyl 4-vinylbenzene sulfonate)(P7)
1H NMR (400 MHz, THF): δ (ppm) = 7.91–7.54 (br, 2H) (sulfonate, ortho); 7.24–6.71 (br, 2H) (sulfonate, meta); 2.17–1.00 (br, 6H) (backbone). Ratio of pentafluorostyrene to pentafluorophenyl sulfonate was 1:3.03. 19F NMR (377 MHz, THF): δ (ppm) = −143.66 to −146.99 (br, 2F) (pentafluorostyrene, ortho); −154.13 to −155.26 (br, 2F) (pentafluorophenyl sulfonate, ortho); −158.42 to −158.97 (br, 1F) (pentafluorostyrene, para); −158.97 to 160.21 (br, 1F) (pentafluorophenyl sulfonate, para); −164.47 to −166.22 (br, 4F) (pentafluorostyrene and pentafluorophenyl sulfonate, meta). GPC (DMF): Mn = 17800 g mol−1, Mw = 21400 g mol−1, Mn/Mw = 1.20.
Poly(octylstyrene) (P8)
The polymer was synthesized vianitroxide mediated polymerization using MAMA-SG1 as alkoxyamine.
1H NMR (300 MHz, CDCl3): δ (ppm) = 7.16–6.60 (br, 2H) (CH aromatic, ortho, next to backbone); 6.60–6.10 (br, 2H) (CH aromatic, meta); 2.67–2.23 (br, 3H) (ar–CH2); 1.64–1.42 (br, 2H) (ar–CH2–CH2); 1.42–0.97 (br, 10H) (CH2 alkyl); 0.97–0.70 (br, 3H) (CH3); 2.15–0.97 (br, 3H) (backbone). GPC (THF): Mn = 23500 g mol−1, Mw = 34300 g mol−1, Mn/Mw = 1.46, yield: 40%.
Poly(octylstyrene)-b-poly(pentafluorophenyl 4-vinylbenzene sulfonate)(P9)
The polymer was synthesized vianitroxide mediated polymerization using (P8) as alkoxyamine.
1H NMR (300 MHz, THF): δ (ppm) = 7.89–7.48 (br, 2H) (sulfonate, ortho); 7.18–6.66 (br, 2H) (octylstyrene, CH aromatic, ortho, next to backbone and sulfonate, meta); 6.66–6.17 (br, 2H) (octylstyrene, CH aromatic, meta); 2.79–2.34 (br, 2H) (ar–CH2); 1.69–1.49 (br, 2H) (ar–CH2–CH2); 1.49–1.05 (br, 10H) (CH2 alkyl); 2.34–1.05 (br, 6H) (backbone); 1.05–0.77 (br, 3H) (CH3). Ratio of octylstyrene to pentafluorophenyl sulfonate was 1:0.35 from NMR. 19F NMR (377 MHz, THF): δ (ppm) = −154.23 to −155.07 (br, 2F) (pentafluorophenyl sulfonate, ortho); −158.87 to −160.08 (br, 1F) (pentafluorophenyl sulfonate, para); −164.94 to −165.87 (br, 2F) (pentafluorophenyl sulfonate, meta). GPC (DMF): Mn = 69200 g mol−1, Mw = 101500 g mol−1, Mn/Mw = 1.47.
Poly(pentafluorophenyl 4-vinylbenzene sulfonate)-b-poly(styrene) (P10)
1H NMR (300 MHz, THF): δ (ppm) = 8.01–7.55 (br, 2H) (benzene sulfonate, ortho); 7.31–6.28 (br, 7H) (styrene, ortho, meta and para; benzene sulfonate, meta); 2.15–1.02 (br, 6H) (backbone). Ratio of styrene to pentafluorophenyl sulfonate was 1:2.17, as calculated from 1H NMR spectrum.
Post-polymerization functionalization
The respective polymer was dissolved in THF and then a threefold excess of amine was added. The solution was then stirred at room temperature or heated to 60 °C overnight (see Table 2). For the conversion with aniline the mixture was heated to 60 °C and stirred for 6 days. The resulting polymer was isolated by precipitation in methanol or n-hexane and was dried in vacuum afterwards. For the conversion with KOH or N,N-dimethylethylenediamine a sevenfold excess of the KOH or amine was used, respectively.Poly(4-vinylbenzene sulfonate) (PA1)
1H NMR (300 MHz, DMSO): δ (ppm) = 7.77–7.22 (br, 2H) (sulfonate, ortho); 6.97–6.10 (br, 2H) (sulfonate, meta); 2.00–0.97 (br, 3H) (backbone). FT-IR (cm−1): 2933 (aryl); 1332 and 1178 (sulfonate).Poly(N-isopropyl 4-vinylbenzene sulfonamide) (PA2 and PA10)
1H NMR (300 MHz, THF): δ (ppm) = 7.84–7.47 (br, 2H) (sulfonate, ortho); 7.08–6.54 (br, 2H) (sulfonate, meta); 6.53–6.18 (br, 1H) (NH); 3.53–3.13 (br, 1H) (isopropyl, CH); 3.13–2.41(br, 3H) (backbone); 1.17–0.77 (br, 6H) (isopropyl, CH3). In the 19F NMR are also small signals of the pentafluorophenyl 4-vinylbenzene sulfonate left. FT-IR (cm−1): 2978 (C–H); 2937 (aryl); 1412 (C–H); 1320 and 1157 (sulfonate).The same reaction was conducted with the polyPVS, but no conversion was achieved. The 1H NMR spectroscopy showed the same signal as for the polyPVS.
Poly(N-dimethylaminoethyl) 4-vinylbenzene sulfonamide) (PA3 and PA12)
1H NMR (300 MHz, THF): δ (ppm) = 7.86–7.44 (br, 2H) (sulfonate, ortho); 7.24–6.55 (br, 2H) (sulfonate, meta); 3.13–2.79 (br, 2H) (CH2 next to sulfonate); 2.56–2.26 (br, 2H) (CH2 next to N–CH3); 2.26–2.00 (br, 6H) (CH3); 2.00–1.35 (br, 3H) (backbone). FT-IR (cm−1): 2929 (aryl); 2805 (–N–CH3); 1461 (C–H); 1324 and 1153 (sulfonate).The kinetic study of the conversion with the polyPVS polymer was not successful. 1H NMR spectra were similar to the starting material.
Poly(N-hexyl 4-vinylbenzene sulfonamide) (PA4)
1H NMR (300 MHz, THF): δ (ppm) = 7.83–7.40 (br, 2H) (sulfonate, ortho); 7.03–6.57 (br, 2H) (sulfonate, meta); 6.57–6.18 (br, 1H) (NH); 2.99–2.69 (br, 2H) (CH2–NH); 2.45–1.01(br, 3H) (backbone); 1.61–1.39 (br, 2H) (CH2–CH2–NH); 1.39–1.01 (br, 4H). In the 19F NMR are very small signals of the polyPFPVS left. FT-IR (cm−1): 2932 (aryl); 2861 (C–H); 1430 (C–H); 1324 and 1153 (sulfonate).Poly(N-cyclopropyl 4-vinylbenzene sulfonamide) (PA5)
1H NMR (300 MHz, THF): δ (ppm) = 7.97–7.44 (br, 2H) (sulfonate, ortho); 7.24–6.40(br, 3H) (sulfonate, meta and NH); 2.41–2.02 (br, 1H) (cyclopropyl, CH); 2.02–1.13 (br, 3H) (backbone); 0.75–0.15 (br, 4H) (cyclopropyl, CH2). FT-IR (cm−1): 3250 (C–H); 2929 (aryl); 1416 (C–H); 1312 and 1153 (sulfonate).Poly((4-vinylbenzene sulfonyl)morpholine) (PA6)
1H NMR (300 MHz, CDCl3): δ (ppm) = 7.78–7.35 (br, 2H) (sulfonate, ortho); 7.04–6.39 (br, 2H) (sulfonate, meta); 4.03–3.31 (br, 4H) (morpholine, CH2, ortho); 3.31–2.63 (br, 4H) (morpholine, CH2, meta); 2.63–1.00 (br, 3H) (backbone). In the 19F NMR are still remaining signals of the polyPFPVS. FT-IR (cm−1): 2922 (aryl); 2857 (C–H); 1454 (C–H); 1347 and 1162 (sulfonate); 1104 (C–O–C).Poly((1,4-vinylbenzene sulfonyl)pyrrolidine) (PA7)
1H NMR (300 MHz, CDCl3): δ (ppm) = 7.88–7.41 (br, 2H) (sulfonate, ortho); 7.01–6.34 (br, 2H) (sulfonate, meta); 3.43–2.91 (br, 4H) (pyrrolidine, CH2 next to NH); 1.90–1.59 (br, 4H) (pyrrolidine, CH2); 1.90–0.96 (br, 3H) (backbone). In the 19F NMR spectrum peaks of the polyPFPVS are still visible. FT-IR (cm−1): 2929 (aryl); 2876 (C–H); 1416 (C–H); 1324 and 1156 (sulfonate).Poly(N,N-diethyl 4-vinylbenzene sulfonamide) (PA8)
1H NMR (300 MHz, THF): δ (ppm) = 7.99–7.40 (br, 2H) (sulfonate, ortho); 7.23–6.43 (br, 2H) (sulfonate, meta); 3.45–2.85 (br, 4H) (diethyl, CH2); 2.38–1.23 (br, 3H) (backbone); 1.23–0.74 (br, 6H) (diethyl, CH3). Partially (∼63%) converted according to the 1H NMR data. FT-IR (cm−1): 2929 (aryl); 1331 and 1152 (sulfonate).Poly(N-phenyl 4-vinylbenzene sulfonamide) (PA9)
1H NMR (300 MHz, DMSO): δ (ppm) = 10.46–8.94 (br, 1H) (NH); 8.10–7.27 (br, 3H) (sulfonate, ortho and N-phenyl, para); 7.27–6.72 (br, 4H) (sulfonate, meta and N-phenyl, ortho); 6.72–93 (br, 2H) (N-phenyl, meta); 2.27–1.00 (br, 3H) (backbone). In the 19F NMR are signals of the polyPFPVS left.Poly(N,N-diisopropyl 4-vinylbenzene sulfonamide) (PA11)
The reaction was conducted with polyPVS, after the conversion the 1H NMR spectroscopy showed only the signals of polyPVS, whereby we conclude that no reaction was observed.Poly(N,N-dimethylaminoethyl) 4-vinylbenzamide) (PA13)
1H NMR (300 MHz, CDCl3): δ (ppm) = 7.94–7.17 (br, 3H) (NH and benzoate, ortho); 6.69–6.07 (br, 2H) (benzoate, meta); 3.73–3.37 (br, 2H) (CH2 next to NH); 2.76–2.42 (br, 2H) (CH2 next to N(CH3)2); 2.42–2.13 (br, 6H) (diethyl, CH3); 1.80–1.03 (br, 3H) (backbone). FT-IR (cm−1): 3312 (aryl and NH); 2935 (C–H); 2825 (N–CH3); 1634 and 1537 (amide), 1456 (C–H).Acknowledgements
R. Forst and K. Köder are acknowledged for their support in the experimental work and M. Mondeshki for the 19F NMR kinetic studies. This research was partly supported by WCU (World Class University) program through the National Research Foundation of Korea funded by the Ministry of Education, Science and Technology (R31-10013).References
- S. Caddick, J. D. Wilden, H. D. Bush, S. N. Wadman and D. B. Judd, Org. Lett., 2002, 4, 2549–2551 CrossRef CAS.
- K. Pudhorn and T. Vilaivan, Tetrahedron Lett., 1999, 40, 5939–5942 CrossRef.
- R. Pandya, T. Murshima, L. Tedeschi and A. G. M. Barrett, J. Org. Chem., 2003, 68, 8274–8276 CrossRef CAS.
- C. Hansch, P. G. Sammes, J. B. Taylor, Comprehensive Medicinal Chemistry, Pergamon Press, Oxford, 1990, vol. 2, ch. 7.1 Search PubMed.
- P. R. Hanson, D. A. Probst, R. E. Robinson and M. Yau, Tetrahedron Lett., 1999, 40, 4761–4764 CrossRef CAS.
- J. D. Wilden, D. B. Judd and S. Caddick, Tetrahedron Lett., 2005, 46, 7637–7640 CrossRef CAS.
- S. Caddick, J. D. Wilden and D. B. Judd, J. Am. Chem. Soc., 2004, 126, 1024–1025 CrossRef CAS.
- S. Caddick, J. D. Wilden and D. B. Judd, Chem. Commun., 2005, 2727–2728 RSC.
- J. D. Wilden, L. Geldeard, C. C. Lee, D. B. Judd and S. Caddick, Chem. Commun., 2007, 1074–1076 RSC.
- J. Bornholdt, K. W. Fjære, J. Felding and J. L. Kristensen, Tetrahedron, 2009, 65, 9280–9284 CrossRef CAS.
- C. C. Lee, R. J. Fitzmaurice and S. Caddick, Org. Biomol. Chem., 2009, 7, 4349–4351 RSC.
- K.-Y. Baek, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5991–5998 CrossRef CAS.
- B. Helms, S. J. Guillaudeu, X. Xie, M. McMurdo, C. J. Hawker and J. M. J. Fréchet, Angew. Chem., Int. Ed., 2005, 44, 6384–6387 CrossRef CAS.
- M. Shirai, A. Kawaue, H. Okamura and M. Tsunooka, Chem. Mater., 2003, 15, 4075–4081 CrossRef CAS.
- C. H. Chen and L. P. Hammett, J. Am. Chem. Soc., 1958, 80, 1329–1331 CrossRef CAS.
- A. K. Sen, S. Roy and V. A. Juvekar, Polym. Int., 2007, 56, 167–174 CrossRef CAS.
- M. Okamura, T. Yamanobe, T. Arai, H. Uehara, T. Komoto, S. Hosoi and T. Kumazaki, J. Mol. Struct., 2002, 602–603, 17–28 CrossRef CAS.
- Y. K. Bhardwaj, H. Mohan, S. Sabharwal and A. B. Majali, Radiat. Phys. Chem., 2000, 58, 373–385 CrossRef CAS.
- H. Kamogawa, A. Kanzawa, M. Kadoya, T. Naito and M. Nanasawa, Bull. Chem. Soc. Jpn., 1983, 56, 762–765 CAS.
- K. Nilles and P. Theato, Eur. Polym. J., 2007, 43, 2901–2912 CrossRef CAS.
- K. Nilles and P. Theato, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 1696–1705 CrossRef CAS.
- K. Nilles and P. Theato, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3683–3692 CrossRef CAS.
- K. T. Wiss, O. D. Krishna, P. J. Roth, K. L. Kiick and P. Theato, Macromolecules, 2009, 42, 3860–3863 CrossRef CAS.
- C. G. Overberger, C. Frazier, J. Mandelman and H. F. Smith, J. Am. Chem. Soc., 1953, 75, 3326–3330 CrossRef CAS.
- B. Y. K. Chong, J. Krstina, T. P. T. Le, G. Moad, A. Postma, E. Rizzardo and S. H. Thang, Macromolecules, 2003, 36, 2256–2272 CrossRef CAS.
- T.-Y. Guo, P. Liu, J.-W. Zhu, M.-D. Song and B.-H. Zhang, Biomacromolecules, 2006, 7, 1196–1202 CrossRef CAS.
- T. P. T. Le, G. Moad, E. Rizzardo, S. H. Thang, PCT Int. Appl., WO98/01478, 1998; Chem. Abstr., 1998, 128, 115390 Search PubMed.
- S. H. Thang, (B.) Y. K. Chong, R. T. A. Mayadunne, G. Moad and E. Rizzardo, Tetrahedron Lett., 1999, 40, 2435–2438 CrossRef CAS.
- G. Moad, Y. K. Chong, A. Postma, E. Rizzardo and S. H. Thang, Polymer, 2005, 46, 8458–8468 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: GPC traces before and after aminolysis, and full 19F NMR spectra during aminolysis. See DOI: 10.1039/c0py00261e |
| ‡ This paper is part of a Polymer Chemistry issue highlighting the work of emerging investigators in the polymer chemistry field. Guest Editors: Rachel O'Reilly and Andrew Dove. |
| This journal is © The Royal Society of Chemistry 2011 |