Orthogonal modification of polymer chain-ends via sequential nitrile oxide–alkyne and azide–alkyne Huisgen cycloadditions†
Ishwar
Singh
a,
Zoya
Zarafshani
b,
Frances
Heaney
*a and
Jean-François
Lutz
*b
aDepartment of Chemistry, National University of Ireland, Maynooth, Co. Kildare, Republic of Ireland. E-mail: mary.f.heaney@nuim.ie; Fax: +353 1 708 3815; Tel: +353 1 708 3802
bFraunhofer Institute for Applied Polymer Research, Geiselbergstrasse 69, Potsdam, 14476, Germany. E-mail: lutz@iap.fhg.de; Fax: +49 331 568 3000; Tel: +49 331 568 1127
First published on 4th October 2010
Abstract
The α- and ω-chain-ends of well-defined polystyrene chains were functionalized using consecutive Huisgen cycloadditions. Firstly, an α-alkyne, ω-azido heterotelechelic polystyrene precursor was synthesized in three steps: (i) atom transfer radical polymerization in the presence of (1,1,1-trimethylsilyl)-2-propynyl 2-bromo-2-isobutyrate, (ii) deprotection of the alkyne function of the initiator and (iii) nucleophilic substitution of the bromine chain-end of the polymer with sodium azide. Afterwards, the chain-ends of the polymer were modified by successive nitrile oxide–alkyne cycloaddition (NOAC) and copper-catalyzed azide–alkyne cycloaddition (CuAAC). 2 Model building blocks were tested for NOAC, while 4 building blocks were studied for CuAAC. In all cases, the orthogonal combination of NOAC and CuAAC allowed the preparation of tailored heterotelechelic polymers.
1. Introduction
The control over polymer end-groups is an important aspect of macromolecular chemistry.1 Indeed, in long homopolymer or copolymer chains, α- and ω-chain-ends are precisely localizable sites for the incorporation of functional moieties (e.g. reactive groups, fluorescent markers, or biological ligands). Thus, over the last years, various methods for controlling polymer chain-ends have been developed. In particular, it has been shown that controlled radical polymerization techniques such as atom transfer radical polymerization (ATRP) or reversible addition–fragmentation chain transfer polymerization (RAFT) are unique platforms for controlling polymer chain-ends.2 In these approaches, the α- or ω-termini can be functionalized using functional initiators, functional control agents or post-modification approaches. In the latter case, it has been lately demonstrated that orthogonal reactions of the “click”-type (e.g. copper-catalyzed azide–alkyne cycloaddition or thiol–ene chemistry) are extremely versatile tools.3 For example, we reported that the copper-catalyzed Huisgen cycloaddition of azides and terminal alkynes is an unprecedented method for modifying the ω-end-groups of ATRP polymer chains.4,5In the present article, a sequential Huisgen cycloaddition strategy was used for modifying the α- and ω-chain-ends of ATRP-made polymer chains (Scheme 1).6 Sequential orthogonal approaches have been recently shown to be interesting pathways for functionalizing hard- and soft-materials.7 Herein, a model polystyrene chain bearing α-alkyne and ω-azido end-groups was modified first by nitrile oxide–alkyne cycloaddition (NOAC) and then by copper-catalyzed azide–alkyne cycloaddition (CuAAC). The former reaction was very recently evidenced to be an efficient tool for precision polymer chemistry.8,9 Indeed, this cycloaddition is regioselective and proceeds in high yields in the absence of any metal catalyst.10 Yet, this reaction is not as popular as CuAAC and therefore the number of commercially available building blocks for NOAC remains limited. Nevertheless, NOAC and CuAAC appear as interesting complementary tools for polymer design.
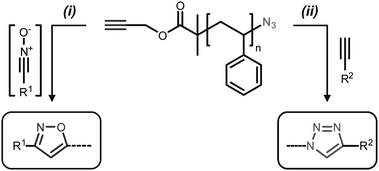 | ||
| Scheme 1 General strategy for the synthesis of well-defined heterotelechelic polystyrene. Experimental conditions: (i) R1CHNOH, NCS, NEt3, CH2Cl2, rt; (ii) CuBr, HMTETA, THF, rt. | ||
2. Experimental part
2.1 Materials
Benzaldehyde oxime 1, 1-naphthaldehyde oxime 5 and the fluorescein derivative 6 were synthesized according to literature protocols.11 All the other reagents were commercially available. The polymer P was synthesized by bulk ATRP of styrene in the presence of (1,1,1-trimethylsilyl)-2-propynyl 2-bromo-2-isobutyrate, copper bromide and pentamethyldiethylenetriamine. The modification steps for preparing the α-alkyne, ω-azido heterotelechelic intermediate were described in a previous publication.122.2 Characterization
Molecular weights and molecular weight distributions were determined by SEC performed at 25 °C in THF (flow rate 1 mL min−1), using four 5 µ-SDV columns (one guard column and three columns of 4 × 103, 3 × 105, and 2 × 106 Å). The detection was performed with a RI (DN-1000, WGE Dr Bures) and a UV/VIS detector (UV 2000, 260 nm). For calibration, linear polystyrene standards (PSS, Germany) were used. Fluorescence spectra were measured using a Jasco Fluorescence Spectrometer FP-6300. 1H NMR spectra were recorded in CDCl3 on a 300 MHz Bruker Avance instrument. 2D-HSQC experiments were performed in CDCl3 on the same instrument operating at 300 MHz (1H)/75 MHz (13C).2.3 General procedure for the nitrile oxide–alkyne cycloaddition (NOAC) step
The nitrile oxide was generated in situ. A stock solution of the required hydroxyimoyl chloride was prepared by dissolving the parent oxime (0.826 mmol) in dichloromethane (6 mL) to which pyridine (20 µL) and N-chlorosuccinimide (0.826 mmol) were added. The mixture was allowed to stir at rt and after 1 h a 2 mL portion of this solution was added to a second flask containing a solution of polymer P (∼200 mg) dissolved in dichloromethane (6 mL). Triethylamine (150 µL) was added and the mixture stirred at rt for 8 h. Further portions of the stock solution (2 mL) were added following 8 and 16 h stirring at rt. The reaction was terminated after a total of 24 h stirring, the solvent was evaporated to ∼1 mL and the product precipitated by addition of cold MeOH, filtered, washed and dried under vacuum.2.4 General procedure for the copper-catalyzed azide–alkyne cycloaddition (CuAAC) step
To a flask containing the isoxazole modified azido polystyrene (P-1 or P-2, ∼50 mg) were added hexamethyltriethylenetetramine (0.136 mmol) and copper bromide (0.066 mmol). The flask was capped with a septum and purged with argon for 2 minutes. In a second flask, a solution of the alkyne (0.066 mmol) in THF (4 mL) was degassed prior to transfer, via a degassed syringe, to the mixture containing the polymer. The mixture was allowed to stir overnight at rt under argon. The solvent was concentrated to approximately 1 mL. Dichloromethane was added and any precipitating salts removed by filtration. Following evaporation of the filtrate to a volume of ∼1 mL, the final product was precipitated by addition of cold MeOH, filtered and dried under vacuum.3. Results and discussion
A well-defined α-alkyne, ω-azido heterotelechelic polystyrene model P was prepared in three steps. The polymer was first synthesized by bulk ATRP in the presence of a radical initiator bearing a trimethylsilyl-protected alkyne function.13 It has been reported that unprotected alkyne-containing initiators can be used in the ATRP of styrene.14 However, side-reactions due to the presence of terminal acetylene functions may occur to a certain extent in radical polymerizations.15 Thus, in the present case, a protected initiator was used in order to minimize chain-end losses. After polymerization and purification, SEC and 1H NMR spectral analysis confirmed the synthesis of a well-defined heterotelechelic precursor (Mn = 3700 g mol−1, Mw/Mn ≈ 1.2). Afterwards, the chain-ends of this intermediate were modified by (i) sodium azide mediated nucleophilic substitution and (ii) TMS-deprotection using tetrabutylammonium fluoride (TBAF). We recently reported that this sequence of chain-end reactions is important.12 Indeed, if the TBAF-deprotection step is performed first, a significant HBr elimination is typically observed at the ω-chain end.Fig. 1A shows the 1H NMR spectrum of the α-alkyne, ω-azido heterotelechelic polystyrene P. A broad chain-end signal can be observed at 3.75–4.25 ppm. Two different types of protons resonate in that region. The signal is due to both the methylene protons located in α- to the alkyne function (protons b in Fig. 1A)16 and to the proton neighboring the azide function, which appears around 4.0 ppm (proton c). This particular signal is indicated by a star in Fig. 1A.17
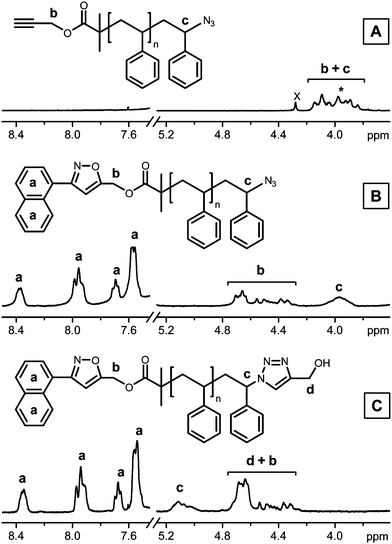 | ||
| Fig. 1 1H NMR spectra (zoom of the regions 8.50–7.45 ppm and 5.27–3.60 ppm) recorded in CDCl3: (A) precursor polymer P before modification; (B) after NOAC modification of the α-chain end with the nitrile oxide derived from 1-naphthaldehyde oxime 2 (Polymer P-2 in Table 1); (C) after CuAAC modification of the ω-chain end with propargyl alcohol 4 (Polymer P-24 in Table 1). | ||
The α-alkyne chain-end of the precursor was first modified by NOAC (Table 1). It has been reported that yields as high as 98% can be obtained by NOAC using equimolar amounts of reactants.8 However, in the present case, an excess of nitrile oxide derivatives was utilized in order to reach nearly quantitative yields. The nitrile oxides were generated in situ from oxime precursors via hydroxyimoyl chloride intermediates. Two different model oximes were used in the present study: benzaldehyde oxime 1 and 1-naphthaldehyde oxime 2 (Scheme 2). Fig. 1B shows the 1H NMR spectrum of a purified polymer after reaction with the nitrile oxide derived from 2. Several new chain-end signals appeared after the NOAC step. For instance, specific signals due to the clicked naphthyl moiety (i.e. protons a in Fig. 1B) can be clearly distinguished in the aromatic region of the spectrum. Moreover, the methylene protons b were significantly shifted downfield (i.e. 4.25–4.85 ppm), thus confirming the formation of an isoxazole ring. Interestingly, this shift also revealed the signal of the proton neighboring the ω-azido end-group (i.e. proton c at 3.8–4.1 ppm). This observation confirms that the azide function remains chemically inert during the NOAC step. Furthermore, the integration values of these various chain-end signals indicated that defined heterotelechelic intermediates were synthesized in all cases (Table 1). A two-dimensional (2D) Heteronuclear Single Quantum Coherence (HSQC) experiment also provided clear evidence of isoxazole formation. Indeed, the CH of the 3,5-disubstituted ring was visualized at 6.41 ppm/104.9 ppm. No evidence for the other regioisomer was found in that spectrum.
 | ||
| Scheme 2 Molecular structure of the model compounds used in the present study for chain-end modification. | ||
| Polymer precursor | Nitrile oxide precursora | Functional alkyneb | Functionalized polymer | Yieldc (%) |
|---|---|---|---|---|
| a Modification of the α-chain end via nitrile oxide–alkyne cycloaddition. b Modification of the ω-chain end via copper-catalyzed azide–alkyne cycloaddition. c Estimated from the 1H NMR spectra of the purified polymers. | ||||
| P | 1 | — | P-1 | >99 |
| P | 2 | — | P-2 | >99 |
| P-1 | — | 3 | P-13 | >99 |
| P-1 | — | 4 | P-14 | >99 |
| P-1 | — | 5 | P-15 | >99 |
| P-2 | — | 4 | P-24 | >99 |
| P-2 | — | 5 | P-25 | >99 |
| P-2 | — | 6 | P-26 | >99 |
In a second step, the ω-end-groups of the polystyrene chains were functionalized by CuAAC (Table 1). Four different alkyne models were tested (structures 3–6 in Scheme 2). In all cases, nearly quantitative chain-end modifications were observed. For instance, Fig. 1C shows the 1H NMR spectrum of the purified polymer P-24 after cycloaddition of P-2 with propargyl alcohol 4. This spectrum clearly indicates that the CuAAC step proceeded in very high yields. As expected, the signal of the proton neighboring the azide function vanished completely and was replaced by a new broad signal at 4.95–5.25 ppm which is characteristic of the methine proton located in the α-position of the formed triazole ring (proton c in Fig. 1C).4 Furthermore, a broad peak corresponding to the methylene protons neighboring the clicked alcohol function (protons d in Fig. 1C) is visible around 4.6 ppm (this particular peak coexists with the signal of protons b). Comparable results were obtained with the functional alkynes 3, 5 and 6 (Table 1). In all cases, the quantitative shift of proton c from 3.8–4.1 ppm to 4.95–5.25 ppm was observed. After CuAAC with 6, the formation of a fluorescent heterotelechelic polymer was also verified by spectrophotometry in dichloromethane. The absorption and emission spectra of the purified polymer P-26 displayed the typical spectroscopic signature of fluorescein.18 It was also verified that the successive NOAC and CuAAC modification steps do not have an effect on the average molecular weight and molecular weight distribution of the polymers. The SEC chromatograms of the modified polymers closely matched that of the precursor P.
4. Conclusion
In conclusion, it was demonstrated that the combination of NOAC and CuAAC is an interesting strategy for controlled modification of the α- and ω-chain-ends of polystyrene chains prepared by ATRP. Various types of heterotelechelic macromolecules could be synthesized using this facile sequential process. This stepwise approach is indeed not limited to polystyrene and can probably be extended to several other types of ATRP-made polymers.Acknowledgements
Financial support from the German Research Foundation (DFG, Sfb 448) and from the Science Foundation of Ireland (Programme code 05/PICA/B838) is gratefully acknowledged.Notes and references
- (a) K. Matyjaszewski and N. V. Tsarevsky, Nat. Chem., 2009, 1, 276–288 Search PubMed; (b) M. Ouchi, T. Terashima and M. Sawamoto, Chem. Rev., 2009, 109, 4963–5050 CrossRef CAS; (c) S. Hilf and A. F. M. Kilbinger, Nat. Chem., 2009, 1, 537–546 Search PubMed.
- (a) V. Coessens, T. Pintauer and K. Matyjaszewski, Prog. Polym. Sci., 2001, 26, 337–377 CrossRef CAS; (b) J.-F. Lutz and K. Matyjaszewski, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 897–910 CrossRef CAS; (c) G. Moad, Y. K. Chong, A. Postma, E. Rizzardo and S. H. Thang, Polymer, 2005, 46, 8458–8468 CrossRef CAS; (d) H. Willcock and R. K. O'Reilly, Polym. Chem., 2010, 1, 149–157 RSC; (e) Y. Xia, N. A. D. Burke and H. D. H. Stöver, Macromolecules, 2006, 39, 2275–2283 CrossRef CAS; (f) W. Jakubowski, B. Kirci-Denizli, R. R. Gil and K. Matyjaszewski, Macromol. Chem. Phys., 2008, 209, 32–39 CrossRef CAS; (g) P. J. Roth, F. D. Jochum, F. R. Forst, R. Zentel and P. Theato, Macromolecules, 2010, 43, 4638–4645 CrossRef CAS.
- (a) H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS; (b) J.-F. Lutz, Angew. Chem., Int. Ed., 2007, 46, 1018–1025 CrossRef CAS; (c) W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2007, 28, 15–54 CrossRef CAS; (d) D. Fournier, R. Hoogenboom and U. S. Schubert, Chem. Soc. Rev., 2007, 36, 1369–1380 RSC; (e) J.-F. Lutz and H. Schlaad, Polymer, 2008, 49, 817–824 CrossRef CAS; (f) A. B. Lowe, Polym. Chem., 2010, 1, 17–36 RSC; (g) A. J. Inglis and C. Barner-Kowollik, Macromol. Rapid Commun., 2010, 31, 1247–1266 CrossRef CAS.
- J.-F. Lutz, H. G. Börner and K. Weichenhan, Macromol. Rapid Commun., 2005, 26, 514–518 CrossRef CAS.
- (a) J.-F. Lutz, H. G. Börner and K. Weichenhan, Macromolecules, 2006, 39, 6376–6383 CrossRef CAS; (b) J.-F. Lutz, H. G. Börner and K. Weichenhan, Aust. J. Chem., 2007, 60, 410–413 CrossRef CAS; (c) S. Pfeifer, Z. Zarafshani, N. Badi and J.-F. Lutz, J. Am. Chem. Soc., 2009, 131, 9195–9197 CrossRef CAS.
- R. Huisgen, Angew. Chem., Int. Ed. Engl., 1963, 2, 565–598 CrossRef.
- (a) P. Kele, G. Mezo, D. Achatz and O. S. Wolfbeis, Angew. Chem., Int. Ed., 2009, 48, 344–347 CrossRef CAS; (b) L. Nurmi, J. Lindqvist, R. Randev, J. Syrett and D. M. Haddleton, Chem. Commun., 2009, 2727–2729 RSC; (c) C. A. DeForest, B. D. Polizzotti and K. S. Anseth, Nat. Mater., 2009, 8, 659–664 CrossRef CAS; (d) N. W. Polaske, D. V. McGrath and J. R. McElhanon, Macromolecules, 2010, 43, 1270–1276 CrossRef CAS; (e) I. Javakhishvili, W. H. Binder, S. Tanner and S. Hvilsted, Polym. Chem., 2010, 1, 506–513 RSC.
- Y. G. Lee, Y. Koyama, M. Yonekawa and T. Takata, Macromolecules, 2009, 42, 7709–7717 CrossRef CAS.
- (a) I. Singh, Z. Zarafshani, J.-F. Lutz and F. Heaney, Macromolecules, 2009, 42, 5411–5413 CrossRef CAS; (b) Y. G. Lee, Y. Koyama, M. Yonekawa and T. Takata, Macromolecules, 2010, 43, 4070–4080 CrossRef CAS; (c) Y. G. Lee, M. Yonekawa, Y. Koyama and T. Takata, Chem. Lett., 2010, 39, 420–421 CrossRef CAS.
- (a) S. Grecian and V. V. Fokin, Angew. Chem., Int. Ed., 2008, 47, 8285–8287 CrossRef CAS; (b) K. S. Wankhede, V. V. Vaidya, P. S. Sarang, M. M. Salunkhe and G. K. Trivedi, Tetrahedron Lett., 2008, 49, 2069–2073 CrossRef CAS; (c) I. Singh, J. S. Vyle and F. Heaney, Chem. Commun., 2009, 3276–3278 RSC; (d) V. Algay, I. Singh and F. Heaney, Org. Biomol. Chem., 2010, 8, 391–397 RSC; (e) I. Singh and F. Heaney, Org. Biomol. Chem., 2010, 8, 451–456 RSC.
- (a) N. Jain, A. Kumar and S. M. S. Chauhan, Tetrahedron Lett., 2005, 46, 2599–2602 CrossRef CAS; (b) H. E. Master, S. I. Khan and K. A. Poojari, Bioorg. Med. Chem., 2005, 13, 4891–4899 CrossRef CAS; (c) S. Derbre, G. Roue, E. Poupon, S. A. Susin and R. Hocquemiller, ChemBioChem, 2005, 6, 979–982 CrossRef CAS.
- M. A. Berthet, Z. Zarafshani, S. Pfeifer and J. F. Lutz, Macromolecules, 2010, 43, 44–50 CrossRef CAS.
- J. A. Opsteen and J. C. M. van Hest, Chem. Commun., 2005, 57–59 RSC.
- (a) N. V. Tsarevsky, B. S. Sumerlin and K. Matyjaszewski, Macromolecules, 2005, 38, 3558–3561 CrossRef CAS; (b) B. A. Laurent and S. M. Grayson, J. Am. Chem. Soc., 2006, 128, 4238–4239 CrossRef CAS.
- (a) S. Pfeifer and J.-F. Lutz, Chem.–Eur. J., 2008, 14, 10949–10957 CrossRef CAS; (b) S. Pfeifer and J.-F. Lutz, Macromol. Chem. Phys., 2010, 211, 940–947 CAS.
- It is interesting to note that the same type of chain-ends leads to relatively sharp signals for poly(acrylate) or poly(methacrylate) polymers. Such signals can be seen for example in ref. 9a or in the following reference: Z. Zarafshani, Ö. Akdemir and J.-F. Lutz, Macromol. Rapid Commun., 2008, 29, 1161–1166 Search PubMed In the case of polystyrene, a broader signal is typically observed. See for example refererence 12a or the following references: A. Hasneen, S. J. Kim and H. J. Paik, Macromol. Res., 2007, 15, 541–546 CrossRef CAS; A. Hasneen, H. S. Han and H.-J. Paik, React. Funct. Polym., 2009, 69, 681–687 Search PubMed It has been proposed that these complex polystyrene chain-end signals are due to stereoisomerism. See the following reference for more information: M. C. Bheda and H. W. Gibson, Macromolecules, 1991, 24, 2703–2708 Search PubMed.
- The typical shape of this signal can be seen in ref. 4.
- It should be noted that these apolar conditions led to a 25–30 nm blue shift as compared to conventional measurements in polar solvents.
Footnote |
| † This paper is part of a Polymer Chemistry issue highlighting the work of emerging investigators in the polymer chemistry field. Guest Editors: Rachel O'Reilly and Andrew Dove. |
| This journal is © The Royal Society of Chemistry 2011 |