Doxorubicin conjugated, crosslinked, PEGylated particles prepared via one-pot thiol-ene modification of a homopolymer scaffold: synthesis and in vitro evaluation†
Lingjiun
Wong
ab,
Maria
Kavallaris
c and
Volga
Bulmus‡
*b
aCentre for Advanced Macromolecular Design (CAMD), School of Chemical Engineering, The University of New South Wales, Sydney 2052, NSW, Australia
bSchool of Biotechnology and Biomolecular Sciences (BABS), The University of New South Wales, Sydney 2052, NSW, Australia. E-mail: volgabulmus@iyte.edu.tr; Fax: +90-232-750 6645; Tel: +90-232-750 6660
cChildren's Cancer Institute Australia for Medical Research, Sydney 2031, NSW, Australia
First published on 20th September 2010
Abstract
Doxorubicin (Dox)-conjugated, poly(ethylene glycol) (PEG) shielded, reversibly crosslinked particles were prepared by a one-pot thiol-ene reaction from a RAFT-synthesized well-defined homopolymer scaffold, poly(pyridyldisulfide ethylmethacrylate) (PPDSM). Dox and PEG modified with maleimide end-groups (mal-Dox and mal-PEG), were covalently attached in one pot to free thiol groups of PPDSM (Mn = 8900 g mol−1 and PDI = 1.18) in the presence of a disulfide reducing agent. ∼ 50% of the total pyridyldisulfide units were conjugated with Dox and PEG (with an equal mol ratio). Particles with an average hydrodynamic diameter of 192 ± 28 nm were observed to form after conjugation. Incubation of these particles with a disulfide reducing agent resulted in the disassociation of the particles. The release of Dox from the particles was pH dependent. The Dox-conjugated PEGylated particles (with a Dox content of 8 wt%) inhibited the viability of human cervical carcinoma cells (HeLa) with an IC50 value of 8 × 10−7 M, determined by an Alamar Blue assay, while the IC50 of free Dox was 1 × 10−7 M. The fluorescence microscopy analyses of the HeLa cells after incubation with the particles for varying times showed that the Dox carried by the particles is taken up efficiently by the cells.
Introduction
Polymeric soft particles are promising vehicles for safer systemic delivery of antitumor drugs via the enhanced permeation and retention (EPR) effect.1–6 To conform with the complex requirements of systemic drug delivery, these vehicles often need to be endowed with a biocompatible, stealth surface such as a PEGylated surface, and chemical-reactive groups for conjugation of therapeutic/targeting agents and locking (crosslinking) the system to maintain integrity during systemic circulation.The preferred method to generate such systems is usually to form crosslinked micelles from amphiphilic block copolymers.7–12 This method requires the synthesis of well-defined amphiphilic copolymers with block sizes suitable to form micelles, the micellization and drug loading, and the crosslinking along with the purification procedures performed after each step. An alternative approach to the generation of soft particles is the use of amphiphilic homopolymer scaffolds modified with the desired entities such as PEG, therapeutic and targeting agents.13–16 This approach potentially provides a facile and versatile synthetic route to the generation of polymeric soft particles for drug delivery.
The synthesis of a thiol-functionalized, well-defined, homopolymer scaffold poly(pyridyldisulfide ethylmethacrylate) (PPDSM) via the reversible addition fragmentation chain transfer polymerization (RAFT) was reported previously in a number of publications.11–14 In these earlier studies, modification of hydrophobic PPDSM with hydrophilic moieties such as a tripeptide and PEG chains via thiol-disulfide exchange and thiol-ene/thiol-acrylate addition reactions led to the formation of spherical nanoparticles in water.13,14 This finding encouraged us to investigate further the use of PPDSM as a scaffold for straightforward preparation of PEGylated nanoparticles as a potential drug delivery vehicle.
In this present study, PEG and a well-known anticancer drug, doxorubicin (Dox) were grafted onto RAFT-synthesized PPDSM polymer via a one-pot thiol-ene click addition reaction (Scheme 1).14 The combination of the RAFT technique and the thiol click chemistry provides an excellent route to the generation of polymeric drug delivery vehicles as it offers effective control over the properties of the final product without the need for harsh reaction conditions and potentially-toxic additives.17–27
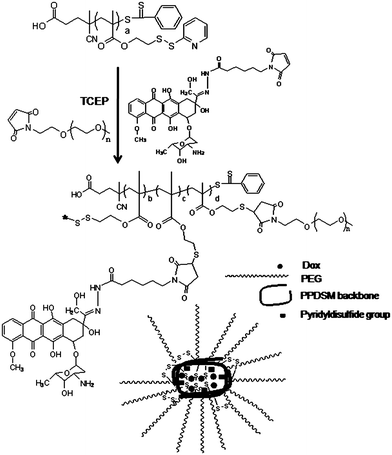 | ||
| Scheme 1 Synthesis of Dox-conjugated, PEGylated, disulfide crosslinked particles from a RAFT-synthesized, thiol-functionalized homopolymer, poly(pyridyldisulfide ethylmethacrylate) (PPDSM). | ||
A maleimide-modified, hydrazide derivative of Dox (mal-Dox) was conjugated to PPDSM in one pot together with maleimide-modified PEG. The derivative of Dox used in the study, has a maleimide group that allows the reaction with the thiol-groups on the polymer via thiol-ene addition, and a carbonyl-reactive hydrazide group that forms an acid-labile hydrazone link between Dox and the polymer. pH - sensitive hydrazone linkages28–30 have been widely used in polymer-drug conjugates to provide drug release at mildly acidic pHs, such as those at tumor and inflammatory tissues, and inside the endocytic vesicles.28,29,31–33 The phase I and pharmacokinetic studies on Dox conjugates linked through a hydrazone bond have revealed that these conjugates have a good safety profile and are able to induce tumor regressions.30
Maleimide-modified PEG (mal-PEG) was grafted to PPDSM via a physiologically-stable thio-ether bond.34 It was shown previously that grafting PEG onto water-insoluble PPDSM homopolymer scaffold via both thiol-acrylate Michael addition and thiol-ene radicalic addition mechanisms leads to the formation of nanoparticles with a PEG shell.14 Moreover, inter- and intra-chain thiol-disulfide exchange reactions take place concurrently with the click addition of PEG to PPDSM due to the presence of free thiol groups along the PPDSM chains.14 Considering these findings, we hypothesised that the one-pot conjugation of Dox and PEG to PPDSM in the presence of an excess reducing agent should yield in one-step Dox-conjugated, PEGylated particles crosslinked via disulfide bonds. Disulfide crosslinked particles are known to be stable in extracellular conditions such as in blood circulation while dissociate at intracellular sites.35–37
Within this study, the one-pot modification of PPDSM with mal-Dox and mal-PEG (Mn 5000 g mol−1) and the physicochemical characterization of the resultant particles by NMR and DLS have been described. The pH-dependent release of Dox from the particles has been investigated using HPLC. The in vitro cytotoxicity and cell uptake of the particles have been assessed using a human cervical carcinoma cell line (HeLa cells) via an Alamar Blue assay and fluorescence microscopy, respectively. The results indicate that Dox-conjugated, PEGylated, reversibly crosslinked particles, generated directly from the RAFT-synthesized homopolymer, show potential for further biological evaluations as a potential Dox delivery system.
Results and discussion
One-pot synthesis of Dox-conjugated, PEGylated, reversibly crosslinked particles from PPDSM scaffold
Mal-Dox and mal-PEG were sequentially grafted to PPDSM (Mn = 8,900 g mol−1 and PDI = 1.18) in a one-pot thiol-ene reaction. PDS functional groups of PPDSM were reduced to thiols by TCEP (approx. 61 mol% of PDS units were targeted) first in the presence of mal-Dox (equivalent to 37 mol% of PDS units). After a 5-hours reaction, mal-PEG (equivalent to 25 mol% of PDS units) along with more TCEP (equivalent to 61% of PDS units) was added to the reaction solution and the resultant solution was further reacted. It is worthwhile noting that starting grafting first with mal-PEG results in low Dox grafting yield. This is possibly due to steric hindrance and solubility problems: the conjugation of the macromolecule chains (PEG) first to the PPDSM backbone might sterically hinder the PDS units from exposing to the Dox. Additionally, Dox is less hydrophilic compared to PEG. Hence the conjugation of Dox to the hydrophobic polymer PPDSM should be more favorable compared to its conjugation to the PEGylated PPDSM which should be a more hydrophilic structure compared to PPDSM.The yield of mal-Dox conjugation was determined by measuring the difference between the concentration of the unconjugated Dox in the reaction mixture aliquot and the concentration of Dox in the feed. The unconjugated Dox concentration in the aliquots, taken from the crude reaction mixture before purification was measured by HPLC using a calibration curve (Figure S4b, ESI), built using standard Dox solutions at known concentrations. By using eqn (1), it was calculated that 66% of the total Dox amount in the feed was conjugated to PPDSM (conjugated to ∼24 mol% of total PDS units on the polymer). It should be noted that upon filtration of crude reaction mixture before injection to HPLC, Dox-conjugated, PEGylated particles are removed from the reaction solution, thus the filtered reaction solution contains only unreacted reagents (mal-Dox, mal-PEG, PPDSM) and by-products (oxidized TCEP, pyridine-2-thione). In order to ensure that all components including pyridine-2-thione (a by-product releasing after the cleavage of pyridyldisulfide units), and active pyridyldisulfide units from PPDSM do not interfere with the HPLC signal at 490 nm, the respective pre-made solutions of each reagent and by-product were injected into HPLC and detected at 490 nm. It was found that only unreacted Dox in the filtered reaction mixture can be detected at 490 nm (Figure S5, ESI). Thus the HPLC method used to measure the unconjugated mal-Dox concentration in the reaction solution allowed the accurate determination of the mal-Dox conjugation yield.
1H-NMR analysis of the PEG- and Dox-conjugated PPDSM provided qualitative data for the PEGylation of PPDSM and proved clearly the presence of Dox and PEG in the product obtained (Figure S6, ESI). Since PEG- and Dox-conjugated PPDSM (PEG is water soluble while PPDSM and doxorubicin are water insoluble) is amphiphilic and possibly crosslinked, PEG and PPDSM signals could not be detected accurately by 1H-NMR in a single spectrum (using varying solvents and solvent mixtures), thus the 1H-NMR analysis did not allow the accurate determination of PEGylation degree of the final product. Thus, the yield for mal-PEG conjugation was determined by comparing the mass of the final product obtained after purification with the initial mass of the reactants in the feed, and found to be approximately 100% (i.e. 25 mol % of total PDS units was conjugated with PEG), consistent with our previous results.14 In summary, the resultant particles had a Dox content of ∼ 8 wt% (∼ 3 mg Dox in 40 mg final product) and a PEG content of ∼ 73 wt% (∼ 29 mg PEG in 40 mg product), and was obtained with 97 wt% overall yield ([40 mg product/41.4 mg total reactants] × 100 = 97%).
Originally water-insoluble PPDSM could be dispersed easily in water after Dox- and PEG- conjugation. The transformation of water-insoluble linear homopolymer PPDSM into a water-dispersable structure further proved that the Dox-conjugated PPDSM was PEGylated. Dynamic light scattering (DLS) measurements in water showed clearly the presence of particles having an average hydrodynamic diameter of 192 ± 28 nm and a PDI of 0.59 ± 0.15 (Fig. 1a). The relatively large PDI indicated the presence of large size particles which suggested the presence of inter-chain hydrophobic interactions and intra- and inter-chain disulfide crosslinking due to the oxidation of free thiol groups formed upon reduction by TCEP. The hydrodynamic diameter of PPDSM conjugated only with PEG (approximately 17 PDS units conjugated with PEG of Mn = 5,000 g mol−1) following the same synthetic protocol was ∼105 ± 6 nm (PDI = 0.20 ± 0.01) in water (data not shown). The smaller hydrodynamic diameter and the narrow size distribution of PPDSM conjugated only with PEG (without Dox) indicated the presence of hydrophobic interactions (and possibly π–π interactions as well) during the particle formation step of Dox-conjugated PEGylated PPDSM, possibly due to the presence of hydrophobic Dox molecules, leading to the formation of bigger particles with larger polydispersity.
 | ||
| Fig. 1 Representative DLS measurements of Dox conjugated PEGylated particles (1 mg ml−1 in water), and after incubation with TCEP for 3, 6 and 12 h, and the average hydrodynamic diameter and polydispersity (PDI) of the particles measured by DLS. | ||
In a separate control experiment performed to investigate the presence of disulfide crosslinking, Dox-loaded, PEGylated nanoparticles (Dox content: 24 mol% of total PDS units, 8 wt%) were incubated with an excess of disulfide reducing agent, TCEP, in water for 3, 6 and 12 h and analyzed by DLS. The result showed that upon incubation with TCEP, the particle size (and PDI) changed from 192 ± 28 nm (PDI = 0.59 ± 0.15) to 127 ± 2 nm (PDI = 0.18 ± 0.01), 59 ± 5 nm (PDI = 0.27 ± 0.0) and then 17 ± 5 nm (PDI = 0.37 ± 0.09) in 3, 6 and 12 h, respectively (Fig. 1b). The decrease in the size of Dox-loaded, PEGylated nanoparticles after incubation with TCEP evidenced that the particles, formed during the one-pot conjugation of Dox and PEG, were crosslinked via disulfide bonds. This feature can give the particles opportunity for intracellular disassociation.38
Previous studies have reported that Dox is not stable in aqueous basic conditions.39,40 Since a basic compound, ethylamine was used as a catalyst for maleimide-thiol addition reaction, it was necessary to check the stability of Dox in the presence of EA. In a control experiment, stability of Dox in the presence of ethylamine (12 mol% of Dox) was monitored by HPLC. Here it should be noted that the ratio of EA to Dox used in the control experiment was higher than the ratio used in the grafting experiment. From the HPLC measurements (Fig. 2), the original Dox structure was retained after incubation with EA for 3 and 30 h, while the Dox sample treated with 0.1 N NaOH solution for 3 h did not show any Dox signal.
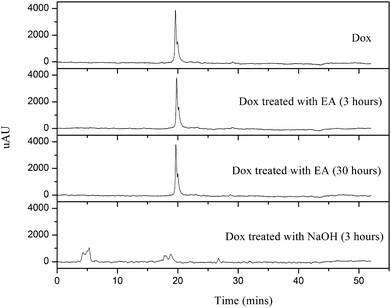 | ||
Fig. 2 HPLC spectra of Dox, Dox incubated with ethylamine (EA, 12 mol% of Dox) for 3 and 30 h in HPLC grade-acetonitrile: water mixture of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :90 (v:v), and Dox incubated in 0.1 N sodium hydroxide solution for 3 h. The initial concentration of Dox for all samples was 44 μM. :90 (v:v), and Dox incubated in 0.1 N sodium hydroxide solution for 3 h. The initial concentration of Dox for all samples was 44 μM. | ||
pH-sensitive release of Dox from PEGylated particles
Macromolecules and particles are taken up by cells via endocytosis mechanism.29,32,41 Endocytic pathway involves acidic membrane-vesicles (endosomes and lysosomes). In addition, some tumor sites have slightly acidic extracellular environment.28,29,31,33 Accordingly, we aimed to synthesize an acid-sensitive polymeric DDS for an antitumor drug, Dox. Dox was conjugated to the particles via an acid-cleavable hydrazone linkage. pH-dependent Dox release from PEGylated particles was evaluated at pH 5.0 and 7.4. The released Dox was monitored by HPLC using a visible light detector at 490 nm. In a control experiment in which the possible interference of the absorbance at 490 nm by the other components present in the release medium (such as pyridine-2-thione and pyridyldisulfide units of PPDSM) was controlled (Figure S5, ESI), it was found that only released Dox can be detected at 490 nm. The release of Dox from the particles was calculated according to the eqn (3). All absorbance values obtained from HPLC measurements were corrected considering appropriate dilution factors for calculation of percent release (Fig. 3). The release of Dox from the particles incubated at pH 5.0 and pH 7.4 for 24 h was 30% and 65% of the total drug loaded, respectively (Fig. 3 and Figure S7 in ESI). In 72 h, the release of Dox at pH 5.0 reached to 80% while the release at pH 7.4 maintained at 30%. There was about less than 20% Dox release at both pHs within the first 5 h, which might be due to unconjugated Dox physically entrapped inside the particles.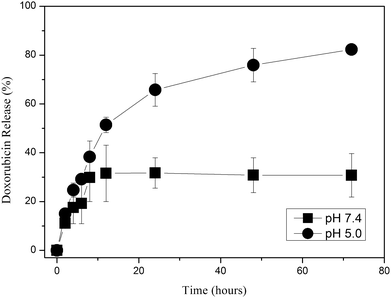 | ||
| Fig. 3 The release of Dox (% of total Dox content) from PEGylated particles (drug content of particles: ∼ 8 wt%). The particles were incubated in phosphate-buffered saline (PBS) pH 7.4 and citrate-phosphate-buffered saline (CPBS) at pH 5.0 separately in duplicate. The release was quantified by HPLC as described in the materials and method section. | ||
Effect of Dox-conjugated PEGylated particles on viability of human cervical carcinoma cell line
The viability of human cervical carcinoma cells (HeLa), after incubation with Dox, Dox conjugated, PEGylated particles (drug content 8 wt%), and PEGylated particles (without Dox) for 72 h was investigated by a CellTiter-Blue® assay according to the manufacturer`s protocol. CellTiter-Blue is a non-toxic, water soluble tetrazolium salt which can be used to monitor the reducing environment of the proliferating cells. The internal environment of the proliferating cells is more reducing than that of non-proliferating cells. The dye can be reduced by metabolic intermediates (NADPH/NADP, FADH/FAD, FMNH/FMN, NADH/NAD) and the reduction is accompanied by a measurable shift in color.42,43 Following the 72 h incubation with the particles, metabolic activity of the cells was detected by the addition of the CellTiter-Blue reagent followed by the fluorescence measurement. The viability of cells was evaluated after treatment with decreasing concentration of Dox (10−5, 10−6, 10−7, 10−8, and 10−9 M) and the cell viabilities were calculated by using eqn (4). Each sample was tested in triplicate. The PEGylated particles without Dox did not inhibit the cell viability over the concentration range tested (Fig. 4). The concentration of Dox required to inhibit cell viability by 50% (IC50) was found to be 1 × 10−7 M (5.4 × 10−8 g ml−1). The IC50 of Dox-loaded PEGylated particles was 8 × 10−7 M Dox (4.3 × 10−7 g Dox/ml or 5.4 × 10−6 g particles/ml) (Fig. 4). Almost eight-fold less IC50 of free Dox, compared to that of Dox loaded particles was observed. Reduced in vitro toxicity of polymer-anticancer drug conjugates/particles compared to free drug is not rare. 10–20-fold decrease has been reported previously by others.3,44,45 The in vitro pharmacokinetics of polymer conjugates/particles is usually limited (by poor diffusion and cell uptake) with respect to free drug. However, the in vivo pharmacokinetics profile can be significantly different than in vitro profile, as polymer conjugates/particles prove to alter biodistribution of drugs minimizing their access to non-targeted healthy sites and alleviate side-effects.1 | ||
| Fig. 4 The average viability of HeLa cells (relative to the control) after the treatment with Dox, PEGylated Dox-conjugated particles (drug content ∼ 8 wt%), PEGylated particles without Dox for 72 h, determined by CellTiter Blue™ assay. The experiments were done in triplicate. The Dox concentration in Dox and Dox-loaded particle samples was varied as follow: 10−5, 10−6, 10−7, 10−8 and 10−9 M. The concentration of the sample that contained only particles without Dox was adjusted to be equivalent to the concentration of the particles within the Dox-loaded particle sample. Accordingly, the particle concentration was varied as follow: ∼6.8 × 10−5 mg ml−1, 6.8 × 10−6 mg ml−1, 6.8 × 10−7 mg ml−1, 6.8 × 10−8 mg ml−1 and 6.8 × 10−9 mg ml−1. The error bars represent the mean standard deviation (±) of data obtained from three different experiments. | ||
Cellular uptake of Dox-conjugated PEGylated particles
HeLa cells were incubated with Dox and Dox-loaded particles (drug content: 8 wt%) for 2 h, 6 h and 24 h, and then imaged by a fluorescence microscope. The Dox concentration was constant in all treatments (10−7 M, 5.4 × 10−5 mg ml−1). Images obtained from the experiments are shown in Fig. 5, 6 and 7 (non-treated cells, and cells after treatment with Dox and Dox-loaded nanoparticles, for 2, 6 and 24 h incubation, respectively). From light microscope images, it was clear that the growth of the cells incubated with Dox and Dox-loaded particles were inhibited significantly as the cell number after treatments was significantly less compared to the cells with no treatment. Both Dox and Dox-loaded PEGylated nanoparticles showed comparable intensity of drug accumulation inside the cells. However, the cell number in samples treated only with Dox was less compared to that in samples treated with Dox-loaded particles, which was consistent with the cell viability assay results. The red fluorescence inside the cells increased gradually from 2 h to 6 h, corresponding to the accumulation and release of Dox in the cells with time. The Image-J software was used to compare the fluorescence intensity of all samples. The cells after treatment for 2-hours with both Dox-only and Dox-loaded particles showed approximately 1.5-fold higher red fluorescence per cell, compared to the cells with no treatment. After 6-hours incubation with Dox-only and Dox-loaded particles, the cells showed approx. 2.5 and 4.6-fold higher red fluorescence, respectively, compared to the cells with no treatment. After 24-hours incubation, the fluorescence intensity per cell decreased slightly (approximately 2.1 and 3.4-fold higher, with free Dox and Dox-loaded particles, relative to the cells with no treatment), along with the decrease in the number of cells, indicating the death of the cells, consistent with the results obtained from the cell viability assay. | ||
| Fig. 5 Fluorescence micrographs of HELA cells (1000 cells/well) (a) without any treatment, (b) after incubation with Dox and (c) after incubation with Dox-loaded PEGylated particles for 2 h. Images were captured at 40X magnification. The concentration of Dox in all three samples was 10−7 M. | ||
 | ||
| Fig. 6 Fluorescence micrographs of HELA cells (1000 cells/well) (a) without any treatment, (b) after incubation with Dox and (c) after incubation with Dox-loaded PEGylated particles, for 6 h. Images were captured at 40X magnification. The concentration of Dox in all three samples was 10−7 M. | ||
 | ||
| Fig. 7 Fluorescence micrographs of HELA cells (1000 cells/well) (a) without any treatment, (b) after incubation with Dox and (c) after incubation with Dox-loaded PEGylated particles for 24 h. Images were captured at 40X magnification. The concentration of Dox in all three samples was 10−7 M. | ||
These results revealed that the take up of Dox carried by the particles was higher than that of free Dox, indicating that the particles are efficiently taken up by HeLa cells and release the active Dox inside the cells. The higher fluorescence intensity of the cells incubated with Dox-loaded particles (for both 6 and 24 h) compared to the cells incubated with Dox-only proved that the fluorescence inside the cells incubated with Dox-loaded particles cannot be attributed to the Dox burst-released from the particles within the first 5-hours (observed in drug release profile in Fig. 3).
Combination of the cell uptake results with the toxicity assay results indicated that the Dox carried by the particles, accumulates more inside the cells, but shows slightly less therapeutic activity than the free Dox. Indeed, in 6 h, the red fluorescence of Dox-loaded particles was observed to accumulate as dots around the nucleus inside the cells (though there was some red fluorescence diffused through nucleus) while the fluorescence in the cells treated with Dox only showed a more diffused pattern. This suggested that the Dox-loaded particles entered into the cells via endocytosis.29,44 The internalization of the Dox-loaded particles by endo/lysosomal vesicles should present an extra diffusion barrier to the released Dox. In addition, although the conjugated Dox is released from the polymer at slightly acidic pH at a higher rate compared to the release at neutral pH, the release rate should be quicker to evade the degradation of some of Dox in endocytic pathway. All these effects should cause the Dox-loaded particles to have a higher IC50 value compared to the free Dox. Nevertheless, the in vitro profile of the drug-loaded particles (compared to free drug) is potentially compensated in vivo by the advantages of the particulate systems such as improved biodistribution and decreased side-effects.
Experimental
Materials
Poly(pyridyldisulfide ethylmethacrylate) (PPDSM; Mn = 8,900 g mol−1 and PDI = 1.18 by GPC) was synthesized and purified according to the procedure reported elsewhere.11,13 The 1H NMR spectrum and GPC chromatogram of PPDSM homopolymer used in this study are shown in Figures S1a and S1b (Electronic Supporting Information, ESI†). Tris(2-carboxyethyl)phosphine (TCEP) hydrochloride (Sigma-Aldrich) was dissolved in deionized water, and neutralized by adding 0.1 M NaOH solution. The resultant solution was freeze-dried and then re-dissolved in absolute methanol and syringe-filtered to remove the salt. The solution was then dried under vacuum to yield the neutralized TCEP. N-[ε-Maleimidocaproic acid] hydrazide (EMCH) (PIERCE) was used as received. Ethylamine (EA) was purchased from Sigma-Aldrich. Doxorubicin (Dox) hydrochloride aqueous solution (50 mg/25 ml concentration) was purchased from DBL (Mayne Pharma Pty Ltd) and carefully neutralized using diluted NaOH solution. The neutralized Dox solution was freeze-dried, re-dissolved in absolute ethanol and then filtered to remove the salt prior to use. The solution was dried under vacuum to yield neutralized Dox. Maleimide derivative of doxorubicin (mal-Dox) that contains a hydrazone bond between the drug and the maleimide functionality was synthesized according to the procedure reported previously (Scheme S1, ESI).11,281H NMR and ESI-MS spectra of mal-Dox are shown in Figures S2a and S2b (ESI), respectively. Maleimide functionalized poly(ethylene glycol) (mal-PEG) (Mn = 5000 g mol−1, purity of maleimide ∼ 41 mol %), was purchased from Fluka. 1H NMR spectrum of mal-PEG is given in Figure S3 (ESI). High purity nitrogen gas (Linde gases, 99.99%) was used to purge the reaction solutions for polymer modifications via thiol-maleimide click reaction. Deuterated chloroform (CDCl3) in silver foil, deuterated dimethyl sulfoxide (DMSO-d6) and deuterated methanol (CD3OD) were purchased from Cambridge Isotope Laboratories and used as received. HPLC-grade acetonitrile and methanol were purchased from Honeywell Burdick and Jackson Brand, respectively. Diethyl ether anhydrous (Univar, analytical grade reagent) was used as received.Dulbecco's modified eagle medium (DMEM) was purchased from GIBCO. Fetal bovine serum (FBS) and Hank's balanced salt solution (HBSS) were obtained from SAFC Biosciences. Trypsin-EDTA was obtained from Thermo Scientific. CellTiter-Blue® cell viability assay reagent was purchased from Promega. Trypan Blue was purchased from Sigma-Aldrich. The cell culture flasks and plates were obtained from NUNC. HeLa cells were kindly donated by Assoc. Prof. Robert Yang at UNSW. Phosphate buffer saline (PBS) pellets were purchased from Sigma-Aldrich.
Analytical techniques
:4 v/v mixture of THF: methanol with a sodium acetate concentration of 0.6 × 10−3 M. Sodium acetate was added to the solvent prior to analysis to ensure that ionization would occur, albeit ionization occurs readily from the sodium content that occurs naturally in the glass. All theoretical molecular weights were calculated using the exact mass for the first peak in any given isotopic pattern. The theoretical isotopic patterns were generated using the XcaliburTM program included with the Thermo Finnigan LCQ Deca ion trap mass spectrometer.
Methods
The crude reaction mixture was then precipitated in anhydrous diethyl ether to remove the solvent used for reaction. The precipitated product was redissolved in absolute ethanol and dialysis was carried out for 2 days using a dialysis tubing of 25,000 g mol−1 molecular weight cut-off. Ethanol was used as an eluent (solvent was changed 4 times a day). The resultant solution was collected and dried under vacuum for at least 5 h. The Dox loading efficiency (wt%) and the drug content of the particles (wt%) were calculated based on the eqn (1) and (2) given below:
| Loading efficiency (%) = [(concentration of drug before reaction – concentration of drug after reaction)/concentration of drug before reaction] × 100 | (1) |
| Drug content (wt%) = [(mass of drug loaded)/mass of final product after drug and PEG conjugation] × 100 | (2) |
Separately, a control experiment was performed to confirm the accuracy of using HPLC to quantify the Dox conjugation to the particles. The control experiment was designed to confirm that the detection of unreacted Dox at 490 nm was not interfered by the presence of the other reaction components in the crude conjugation reaction mixture. The solutions of PPDSM (1 mg, Mn = 8,900 g mol−1 and PDI = 1.18, dissolved in 1.6 ml HPLC-grade DMF), pyridine-2-thione (1 mg diluted in 1.6 ml in HPLC-grade DMF) and mal-Dox (20 μg in 1 ml HPLC grade-acetonitrile: water mixture of 10:90) were prepared separately. The solvents used to dissolve the samples were chosen to be the best solvents for the samples to yield a good detection by HPLC if there is any absorption at 490 nm. The samples were filtered using a 0.45 μm PTFE grade hydrophilic membrane filter and injected into HPLC. The fractions were detected using a UV-visible absorbance detector at 490 nm.
Briefly, neutralized Dox (1.84 μmoles) was dissolved in HPLC grade methanol (160 μL). Approximately EA (0.22 μmoles in 14 μL methanol, 12 mol% of Dox) was added into the solution. After 3 and 30 h, aliquots of 7 μl (0.07 μmol Dox) were taken and diluted to 1.6 ml with HPLC grade-acetonitrile: water (10:90) mixture. The final Dox concentration in the diluted aliquot was 44 μM (25 μg ml−1). The final solution was injected into HPLC to investigate the degradation of Dox in basic environment. The spectra obtained were compared with the HPLC spectra of both untreated Dox and Dox degraded by the treatment with a concentrated sodium hydroxide (NaOH) solution (0.1 N NaOH solution) for 3 h.39,40
| Drug Release (%) at any time = [concentration of drug released at any time/concentration of drug released after complete hydrolysis] × 100 | (3) |
| Cell viability (%) = [(Fsample well– Fblank well)/(Fpositive control well−Fblank well)] × 100 mol % | (4) |
Conclusion
In conclusion, we have exploited a new synthetic approach to prepare in one-step doxorubicin-conjugated, PEGylated and reversibly crosslinked particles. Overall the results indicate that simply grafting Dox and PEG to the reactive, well-defined homopolymer scaffold in one pot via thiol-ene click reaction can be a straightforward route to the generation of a soft particulate system for potential delivery of Dox. The preliminary biological evaluation of the generated particles via this new approach shows the potential of the system for further testing as a delivery system for Dox.Acknowledgements
We thank the Australian Research Council for Discovery Research Grant (DP0664805). We also acknowledge Dr Adelle Sasha, Dr Donald Thomas and Dr James Hook from the NMR Spectrometry Facilities at UNSW. In addition we thank Mrs. Nazli Naimi and Mr. Karthikeyan Gunasekaran (BABS, UNSW) for their help with the cell culture experiments.References
- R. Duncan, Nat. Rev. Cancer, 2006, 6, 688–701 CrossRef CAS.
- L. Andersson, J. Davies, R. Duncan, P. Ferruti, J. Ford, S. Kneller, R. Mendichi, G. Pasut, O. Schiavon, C. Summerford, A. Tirk, F. M. Veronese, V. Vincenzi and G. Wu, Biomacromolecules, 2005, 6, 914–926 CrossRef CAS.
- P. Chytil, T. Etrych, C. Konak, M. Sirova, T. Mrkvan, J. Boucek, B. Rihova and K. Ulbrich, J. Controlled Release, 2008, 127, 121–130 CrossRef CAS.
- H. Maeda, G. Y. Bharate and J. Daruwalla, Eur. J. Pharm. Biopharm., 2009, 71, 409–419 CrossRef CAS.
- H. Maeda, L. W. Seymour and Y. Miyamoto, Bioconjugate Chem., 1992, 3, 351–362 CrossRef CAS.
- J. H. Kim, Y. S. Kim, K. Park, S. Lee, H. Y. Nam, K. H. Min, H. G. Jo, J. H. Park, K. Choi, S. Y. Jeong, R. W. Park, I. S. Kim, K. Kim and I. C. Kwon, J. Controlled Release, 2008, 127, 41–49 CrossRef CAS.
- K. L. Wooley, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 1397–1407 CrossRef CAS.
- M. H. Stenzel, Chem. Commun., 2008, 3486–3503 RSC.
- C. L. McCormick, B. S. Sumerlin, B. S. Lokitz and J. E. Stempka, Soft Matter, 2008, 4, 1760–1773 RSC.
- Y. Chan, T. Wong, F. L. Byrne, M. Kavallaris and V. Bulmus, Biomacromolecules, 2008, 9, 1826–1836 CrossRef CAS.
- Z. Jia, L. Wong, T. P. Davis and V. Bulmus, Biomacromolecules, 2008, 9, 3106–3113 CrossRef CAS.
- Z. Jia, J. Liu, C. Boyer, T. P. Davis and V. Bulmus, Biomacromolecules, 2009, 10, 3253–3258 CrossRef CAS.
- L. Wong, C. Boyer, Z. Jia, H. M. Zareie, T. P. Davis and V. Bulmus, Biomacromolecules, 2008, 9, 1934–1944 CrossRef CAS.
- L. Wong, S. Sevimli, H. M. Zareie, T. P. Davis and V. Bulmus, Macromolecules, 2010, 43, 5365–5375 CrossRef CAS.
- T. S. Kale, A. Klaikherd, B. Popere and S. Thayumanavan, Langmuir, 2009, 25, 9660–9670 CrossRef CAS.
- Y. J. Kim, D. Shin, D. R. Vutukuri, E. N. Savariar, S. Y. Kim, S. Thayumanavan and T. P. Russell, Macromolecules, 2007, 40, 4267–4275 CrossRef CAS.
- V. S. Khire, T. Y. Lee and C. N. Bowman, Macromolecules, 2007, 40, 5669–5677 CrossRef.
- A. E. Rydholm, C. N. Bowman and K. S. Anseth, Biomaterials, 2005, 26, 4495–4506 CrossRef CAS.
- K. L. Killops, L. M. Campos and C. J. Hawker, J. Am. Chem. Soc., 2008, 130, 5062–5064 CrossRef CAS.
- L. M. Campos, K. L. Killops, R. Sakai, J. M. J. Paulusse, D. Damiron, E. Drockenmuller, B. W. Messmore and C. J. Hawker, Macromolecules, 2008, 41, 7063–7070 CrossRef CAS.
- B. Yu, J. W. Chan, C. E. Hoyle and A. B. Lowe, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3544–3557 CrossRef CAS.
- A. B. Lowe, Polym. Chem., 2010, 1, 17–36 RSC.
- C. Boyer, A. Granville, T. P. Davis and V. Bulmus, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3773–3794 CrossRef CAS.
- Z. Jia, J. Liu, T. P. Davis and V. Bulmus, Polymer, 2009, 50, 5928–5932 CrossRef CAS.
- C. W. Chang, E. Bays, L. Tao, S. N. S. Alconcel and H. D. Maynard, Chem. Commun., 2009, 3580–3582 RSC.
- A. W. York, S. E. Kirkland and C. L. McCormick, Adv. Drug Delivery Rev., 2008, 60, 1018–1036 CrossRef CAS.
- A. E. Smith, X. Xu and C. L. McCormick, Prog. Polym. Sci., 2010, 35, 45–93 CrossRef CAS.
- D. Willner, P. A. Trail, S. J. Hofstead, H. D. King, S. J. Lasch, G. R. Braslawsky, R. S. Greenfield, T. Kaneko and R. A. Firestone, Bioconjugate Chem., 1993, 4, 521–527 CrossRef CAS.
- M. Prabaharan, J. J. Grailer, S. Pilla, D. A. Steeber and S. Gong, Biomaterials, 2009, 30, 5757–5766 CrossRef CAS.
- C. Unger, B. Haering, M. Medinger, J. Drevs, S. Steinbild, F. Kratz and K. Mross, Clin. Cancer Res., 2007, 13, 4858–4866 CrossRef CAS.
- E. S. Lee, K. Na and Y. H. Bae, Nano Lett., 2005, 5, 325–329 CrossRef CAS.
- V. Bulmus, Aust. J. Chem., 2005, 58, 411–422 CrossRef CAS.
- R. A. Cardone, V. Casavola and S. J. Reshkin, Nat. Rev. Cancer, 2005, 5, 786–795 CrossRef CAS.
- M. Koizumi, K. Endo, M. Kunimatsu, H. Sakahara, T. Nakashima, Y. Kawamura, Y. Watanabe, T. Saga and J. Konishi, Cancer Res., 1988, 48, 1189–1194 CAS.
- S. Bauhuber, C. Hozsa, M. Breunig and A. Goepferich, Adv. Mater., 2009, 21, 3286–3306 CrossRef CAS.
- G. Cavallaro, M. Campisi, M. Licciardi, M. Ogris and G. Giammona, J. Controlled Release, 2006, 115, 322–334 CrossRef CAS.
- S. Cerritelli, D. Velluto and J. A. Hubbell, Biomacromolecules, 2007, 8, 1966–1972 CrossRef CAS.
- V. Bulmus, M. Woodward, L. Lin, N. Murthy, P. Stayton and A. Hoffman, J. Controlled Release, 2003, 93, 105–120 CrossRef CAS.
- M. Fiallo, A. Laigle, M. N. Borrel and A. Garnier-Suillerot, Biochem. Pharmacol., 1993, 45, 659–665 CrossRef CAS.
- J. H. Beijnen, O. A. G. J. Van der Houwen and W. J. M. Underberg, Int. J. Pharm., 1986, 32, 123–131 CrossRef CAS.
- P. Watson, A. T. Jones and D. J. Stephens, Adv. Drug Delivery Rev., 2005, 57, 43–61 CrossRef CAS.
- J. O'Brien, I. Wilson, T. Orton and F. Pognan, Eur. J. Biochem., 2003, 267, 5421–5426 CrossRef.
- Y. Chan, V. Bulmus, H. M. Zareie, F. L. Byrne, L. Barner and M. Kavallaris, J. Controlled Release, 2006, 115, 197–207 CrossRef CAS.
- R. Tomlinson, J. Heller, S. Brocchini and R. Duncan, Bioconjugate Chem., 2003, 14, 1096–1106 CrossRef CAS.
- F. M. Veronese, O. Schiavon, G. Pasut, R. Mendichi, L. Andersson, A. Tsirk, J. Ford, G. Wu, S. Kneller, J. Davies and R. Duncan, Bioconjugate Chem., 2005, 16, 775–784 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H NMR spectrum and GPC of the RAFT synthesized PPDSM, 1H-NMR spectrum and ESI-MS spectrum of mal-Dox, 1H-NMR spectrum of mal-PEG, HPLC traces of standard Dox solutions at increasing concentrations and the calibration curve built by HPLC using standard Dox solutions having known concentration, HPLC traces for filtered crude reaction mixture of PPDSM conjugation with mal-Dox and mal-PEG, pure mal-Dox solution, pure PPDSM solution, pure pyridine-2-thione solution, 1H NMR spectrum of Dox-conjugated and PEGylated PPDSM, representative HPLC traces of Dox released from the PEGylated nanoparticles. See DOI: 10.1039/c0py00256a |
| ‡ Present address: Izmir Institute of Technology, Department of Chemical Engineering, Gulbahce, Urla, Izmir 35430 Turkey. |
| This journal is © The Royal Society of Chemistry 2011 |