Linear dendritic polymeric amphiphiles with intrinsic biocompatibility: synthesis and characterization to fabrication of micelles and honeycomb membranes†‡
Pontus
Lundberg
a,
Marie Valérie
Walter
a,
Maria Isabel
Montañez
ab,
Daniel
Hult
a,
Anders
Hult
a,
Andreas
Nyström
c and
Michael
Malkoch
*a
aFiber and Polymer Technology, Royal Institute of Technology, Teknikrinen 56-58, SE-10044, Stockholm, Sweden. E-mail: malkoch@kth.se; Fax: +4687908283; Tel: +4687908768
bF-IMABIS-Carlos Haya Hospital, Research Laboratory, 29009 Malaga, Spain
cKarolinska Institutet, Department of Neuroscience, Retzius väg 8, S-171 77, Stockholm, Sweden
First published on 18th October 2010
Abstract
Linear dendritic hybrid materials enable a range of architectural variations which offers novel possibilities in the tailoring of polymeric materials. In this study dendrons based on the 2,2-bis(methylol)propionic acid (bis-MPA) building block, bearing click chemistry moieties in the core and peripheral hydroxyl functionalities, have been used as macroinitiators for ring opening polymerization of ε-caprolactone. A library of star branched polymers with poly(ε-caprolactone) chains was initially constructed using dendrons up to 4th generation. In a second step, the popular CuAAC or thiol–ene click reaction was efficiently used to attach poly(ethylene glycol) chains of different lengths to the core. Potential applications of the resulted amphiphilic linear dendritic hybrids were investigated. Both self-assembled micelles loaded with doxorubicin anticancer drug and ordered honeycomb membranes with enhanced surface area were successfully fabricated and characterized.
Introduction
Linear dendritic hybrid materials is a class of architecturally advanced materials that today are receiving increasing attention. This is due to the possibility of joining different polymeric architectures into evermore complex structures using branched dendritic structure with a defined number of branching units. From their initial introduction in which linear polymers were simply end-functionalised with dendritic wedges, an array of structural variations have been reported on.1–3 It is foreseen that these types of materials can be used in a wide range of applications, such as nano-reactors, in catalysis applications, as well as in a range of biomedical applications including drug delivery.1A facile strategy for preparing sophisticated linear dendritic hybrid materials is the combination of ring-opening polymerization (ROP) and click chemistry. This strategy was pursued by Hua et al.4 employing the end-groups of PAMAM® type dendrons as macroinitiators for the ROP of ε-caprolactone, and the alkyne functional core for the attachment of poly(ethylene glycol) (PEG) through click chemistry.
When designing materials for biomedical applications, the biocompatible and degradable nature of the carrier is of great importance. The biocompatible properties of poly(caprolactone) (PCL)5 and PEG6 are well known, and the polymers are extensively used in research concerning biomedical materials. Of the many available dendritic materials reported in the literature, the commercially available dendrimers and dendrons based on 2,2-bis(methylol)propionic acid (bis-MPA) building block have proven to be a leading candidate for the preparation of non-toxic and non-offensive biocompatible materials used in in vivo applications.7–13 These dendritic materials are therefore ideal candidates for the preparation of linear dendritic hybrid materials, supported by our earlier work, that has shown that bis-MPA based dendritic polymers are efficient macroinitiators for ROP.14
The click concept, as postulated by Sharpless and coworkers,15 has been exploited to a great extent within the polymer community due to the unprecedented control and versatility that it offers in preparing architecturally advanced materials. The most popular click reaction is the copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC)16,17 as seen by the large number of recent reviews on the subject.18–29 The most recent addition to the family of click reaction is the free radical initiated thiol–ene chemistry,30–35 that has been raised as a popular metal-free alternative to the CuAAC system. This is of great interest since the use of copper and other heavy metals is an issue of concern if aiming for biomedical applications.
A highly desirable application for amphiphilic linear dendritic hybrid materials is their use as micellar vehicles for drug delivery. Micelles provide increased solubility of hydrophobic drugs. Additional benefits that can be seen using micelles as drug delivery system (DDS) include: improved bioavailability, reduced systemic toxicity, enhanced permeability across physiological barriers, as well as improved biodistribution. The formation of nano-sized micelles is a spontaneous process for amphiphilic block-copolymers and occurs when the polymer concentration is above the critical micelle concentration (CMC). The CMC is one of the most important properties of a self-assembling amphiphilic material. Especially when aiming for applications as a systemically injected drug delivery vehicle, where the injected drug loaded micelles will immediately be diluted by the blood upon administration. If the CMC is too high the micelles will dissociate when diluted, releasing the drug prematurely and no benefit of controlled release is attained. Therefore, the CMC should ideally be in the low mM region or lower.36 Block-copolymers with a predominant hydrophobic block are also intriguing for the fabrication of ordered honeycomb membranes. Since the discovery of hydrophobic polymers ability to form ordered porous structure via spontaneous precipitation on water droplets,37 several polymeric architectures have been shown to form such structure.38
In this study, Scheme 1, we exploit peripheral hydroxylated bis-MPA dendrons of different generations (G0–G4) as functionalized macroinitiators for the construction of sophisticated amphiphilic dendritic-linear hybrid materials with hydrophobic PCL at the chain-ends and a single strain of hydrophilic PEG at the core. ROP was combined with CuAAC and thiol–ene click chemistry to generate inherently biocompatible and physiologically biodegradable scaffolds. These were utilized for the fabrication of drug loaded micelles as well as the development of isoporous membranes.
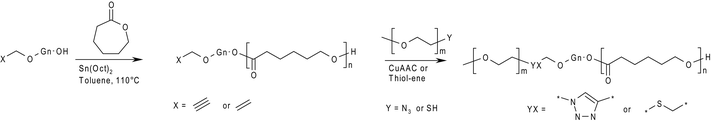 | ||
| Scheme 1 General procedure for the synthesis of linear dendritic hybrid block-copolymers by ROP and CuAAC or thiol–ene click chemistry. | ||
Experimental
Materials
CH2Cl2 (HPLC), diethyl ether (analytical), and MeOH (analytical) were acquired from Fisher Scientific. Acetone (anhydrous), pyridine (AnalR NormaPur), and THF (unstabilized, HPLC) were acquired from VWR. DMSO (for synthesis), Et3N (for synthesis), n-heptane (EMSURE), EtOAc (EMPROVE exp), and toluene (Seccosolv) were acquired from Merck. KOH and benzene (Pro Analysi) were acquired from Acros. Poly(ethylene glycol)s (PEGs), pyrene (puriss), CaH2 (95%), NaN3, N,N-diisopropylethylamine (99.5%) (DIPEA), and propargyl alcohol (≥99%) were acquired from Sigma-Aldrich. Allyl bromide (99%), acetone (spectrophotometric grade), and DOWEX were acquired from Alfa Aeaser. Bis-MPA was supplied by Perstorp and 2,2-dimethoxy-1,2-di(phenyl)ethanone (DMPA) was supplied by CIBA. ε-Caprolactone (Acros) was distilled over CaH2 and stored over 4 Å molecular sieves under Ar (g). Tin(II) 2-ethylhexanoate (95%) (SnOct2) was dried using 4 Å molecular sieves in a solution of toluene prior to use. Acetonide protected bis-MPA anhydride was synthesized as described previously.13Cu(PPh3)3Br was synthesized as described previously.39 The alkyne functional dendrons (alk-Gn) were purchased from Polymer Factory Sweden AB and the synthesis of allyl functional dendrons (all-Gn) is found in the ESI†. PEG2k–N3 and PEG5k–N33 were synthesized as reported earlier.40Methods
1H-NMR and 13C-NMR spectroscopy were performed on a Bruker AM 400 MHz NMR using CDCl3 as a solvent and with TMS as internal reference. MALDI-TOF MS was performed on a Bruker Uniflex MALDI-TOF MS with SCOUT-MTP Ion Source (Bruker Daltonics) equipped with a N2-laser (337 nm), a gridless ion source and reflector design. 9-Nitroanthracene or dihydroxybenzoic acid was used as matrix with added sodium trifluoroacetate. The obtained spectra were analyzed using FlexAnalysis (Bruker Daltonics). SEC using THF as mobile phase (1 ml min−1) at 35 °C was performed on a Viscotek TDA model 301 equipped with 2 T5000 columns, a VE5200 GPC autosample, a VE1121 GPC solvent pump and a VE5710 GPC degasser (all from Viscotek/Malvern). Conventional calibration using linear poly(styrene) standards was used and the samples were evaluated using OmniSEC 4.5 software. FT-IR spectra were collected using a Perkin-Elmer Spectrum 2000 FT-IR equipped with a MKII Golden Gate™ single reflection ATR System from Specac Ltd (16 scans). DSC was performed on a Mettler Toledo DSC820 using a heating/cooling rate of 10 °C min−1 under N2 atmosphere. DLS experiments were performed on a Malvern ZEN 3600 Zetasizer Nano ZS operating at 633 nm and 25 °C. TEM analysis was performed on a JEM-2010/INCA OXROD TEM (JEOL/OXFORD) at a 200 kV accelerating voltage. Samples were prepared by applying micelle solutions to plasma etched carbon coated copper grid. Using filter paper excess solution was wicked away and the samples were stained using uranyl acetate (saturated solution in H2O). FE-SEM images of gold sputtered isoporous membranes were acquired using a Hitachi S-4300 FE-SEM. Fluorescence spectroscopy was performed on a Varian Cary Eclipse. UV-Vis spectroscopy was performed on a Varian Cary 300 Bio.Polymerization of ε-CL using dendrons with alkyne or allyl functional cores as initiators (alk–Gn–PCL or all–Gn–PCL)
Dendrons of generation 0 to 4 with alkyne or allyl functional cores were used as macroinitiators for ROP of ε-CL using Sn(Oct)2 as catalyst in toluene at 110 °C. The targeted degrees of polymerization (DPs) were set to be reached at 75% monomer conversions. A typical example is as follows: a flame dried 100 ml round bottom flask equipped with a stir bar was charged with 189 mg (1.10 mmol) of alk–G1–OH. The flask was sealed with a septum and 2 ml of dry toluene were added. After heating to 110 °C, toluene was removed under vacuum for a period of 20 min and refilled with argon, 20 ml of dry toluene and 19.0 ml of ε-CL (175 mmol). Finally, 0.22 ml of a 1.00 mmol ml−1Sn(Oct)2 stock solution (0.22 mmol) was added. The reaction was allowed to proceed until a conversion of 75% had been reached (usually 2–3 h), as observed by 1H-NMR. The reaction vessel was removed from the oil bath, diluted with 40 ml of CH2Cl2, and precipitated in 1.5 l of MeOH. The white precipitate was subsequently filtered off and dried under vacuum. Yield 14.47 g (92%). 1H-NMR (CDCl3) δ(ppm) = 1.27 (s, 3H, H3C–C), 1.35–1.42 (m, backbone, –CH2–CH2–CH2–), 1.61–1.69 (m, backbone, –CH2–CH2–CH2–), 2.31 (t, backbone, J = 16 Hz, –CH2–C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)–), 3.64 (t, 4H, J = 12 Hz, –CH2–OH), 4.06 (t, backbone, J = 12 Hz, –CH2–O–), 4.24 (m, 4H, –CH2–O– dendron), 4.71 (d, 2H, J = 4 Hz,
O)–), 3.64 (t, 4H, J = 12 Hz, –CH2–OH), 4.06 (t, backbone, J = 12 Hz, –CH2–O–), 4.24 (m, 4H, –CH2–O– dendron), 4.71 (d, 2H, J = 4 Hz, ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–CH2–O–). IR spectra of alk–Gn–PCL and all–Gn–PCL materials are found in the ESI†.
C–CH2–O–). IR spectra of alk–Gn–PCL and all–Gn–PCL materials are found in the ESI†.
Synthesis of thiol functional PEG (PEG2k–SH)
To a 250 ml round bottom flask, equipped with a stir bar, 20.0 g (10.0 mmol) of PEG2k, 5.30 g (50.0 mmol) of mercaptopropionic acid, 0.20 g (1.00 mmol) of p-toluenesulfonic acid and 100 ml of toluene were added. Using a Dean–Stark apparatus the reaction was subsequently heated to 130 °C and allowed to proceed overnight. The product was isolated by precipitation twice in 2 l of diethyl ether followed by filtration and drying under vacuum. Yield 18.4 g (88%). 1H-NMR (CDCl3) δ(ppm) = 1.68 (t, 1H, J = 16 Hz, –SH), 2.66–2.78 (m, 4H, –C(O)–CH2–CH2–SH), 3.37 (s, 3H, O–CH3), 3.64–3.69 (m, backbone, CH2–CH2–O–), 4.26 (t, 2H, J = 8 Hz, –CH2–O–C(O)–). FT-IR spectra can be seen in the ESI†.
Synthesis of amphiphilic linear dendritic hybrid block-copolymers using CuAAC click chemistry (PEG–Gn–PCL)
Generally, CuAAC reactions for the formation of amphiphilic linear dendritic hybrids had a alk-Gn-PCL![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PEG-N3:Cu(PPh3)3Br:DIPEA feed ratio of 1:1.2:0.5:0.25. A typical example is as follows: a 10 ml round bottom flask was charged with 1.00 g (0.081 mmol) of alk–G1–PCL60 and 0.20 g (0.099 mmol) of PEG2k–N3 and dissolved in 5 ml of THF. This was followed by the addition of 26 mg of DIPEA and 38 mg of Cu(PPh3)3Br. The reaction mixture was subsequently heated to 40 °C and the reaction was allowed to proceed overnight. Using 1H-NMR the conversion of the alkyne group was monitored. When complete conversion of the alkyne group had been reached the product was isolated by precipitation in 100 ml cold MeOH followed by filtration and drying under vacuum. Yield 1.1 g (92%). 1H-NMR (CDCl3) δ(ppm) = 1.22 (s, 3H, dendron –CH3), 1.35–1.42 (m, PCL backbone –CH2–CH2–CH2–), 1.61–1.69 (m, PCL backbone –CH2–CH2–CH2–), 2.31 (t, PCL backbone –CH2–C(O)–), 3.38 (s, 3H, –O–CH3) 3.62–3.65 (m, PEG backbone –CH2–CH2–O–), 4.06 (t, J = 12 Hz, PCL backbone –CH2–O–), 4.20–4.24 (m, dendron –CH2–O–), 4.55 (t, 2H, J = 8 Hz –CH2–triazole) 5.25 (2H, triazole–CH2–O–) 7.79 (s, 1H, N–CH). FT-IR spectra of all the PEG–Gn–PCL materials can be seen in the ESI†.
:PEG-N3:Cu(PPh3)3Br:DIPEA feed ratio of 1:1.2:0.5:0.25. A typical example is as follows: a 10 ml round bottom flask was charged with 1.00 g (0.081 mmol) of alk–G1–PCL60 and 0.20 g (0.099 mmol) of PEG2k–N3 and dissolved in 5 ml of THF. This was followed by the addition of 26 mg of DIPEA and 38 mg of Cu(PPh3)3Br. The reaction mixture was subsequently heated to 40 °C and the reaction was allowed to proceed overnight. Using 1H-NMR the conversion of the alkyne group was monitored. When complete conversion of the alkyne group had been reached the product was isolated by precipitation in 100 ml cold MeOH followed by filtration and drying under vacuum. Yield 1.1 g (92%). 1H-NMR (CDCl3) δ(ppm) = 1.22 (s, 3H, dendron –CH3), 1.35–1.42 (m, PCL backbone –CH2–CH2–CH2–), 1.61–1.69 (m, PCL backbone –CH2–CH2–CH2–), 2.31 (t, PCL backbone –CH2–C(O)–), 3.38 (s, 3H, –O–CH3) 3.62–3.65 (m, PEG backbone –CH2–CH2–O–), 4.06 (t, J = 12 Hz, PCL backbone –CH2–O–), 4.20–4.24 (m, dendron –CH2–O–), 4.55 (t, 2H, J = 8 Hz –CH2–triazole) 5.25 (2H, triazole–CH2–O–) 7.79 (s, 1H, N–CH). FT-IR spectra of all the PEG–Gn–PCL materials can be seen in the ESI†.
Synthesis of amphiphilic linear dendritic hybrid block-copolymers using thiol–ene click chemistry (TE–PEG2k–G1–PCL60 and TE–PEG2k–G4–PCL30)
Generally, thiol–ene click reactions for the formation of amphiphilic linear dendritic hybrids had a all–Gn–PCL:PEG–SH feed ratio of 1:3. A typical example is as follows: a 5 ml round bottom flask equipped with a stir bar was charged with 500 mg (34 µmol) of all–G1–PCL60 and 210 mg (100 µmol) of PEG2k–SH. After the complete dissolution of the materials in THF (unstabilised), a catalytic amount of the photoinitiator DMPA was added and the solution was purged with Ar (g) for 2 min followed by exposure to UV for 60 min using a high pressure mercury lamp (Oriel). 1H-NMR (CDCl3) δ(ppm) = 1.26 (s, 3H, dendron –CH3), 1.35–1.42 (m, PCL backbone –CH2–CH2–CH2–), 1.61–1.69 (m, PCL backbone –CH2–CH2–CH2–), 2.31 (t, PCL backbone –CH2–C(O)–), 3.38 (s, 3H, –O–CH3) 3.62–3.65 (m, PEG backbone –CH2–CH2–O–), 4.06 (t, J = 12 Hz, PCL backbone –CH2–O–), 2.58–2.82 (m, 6H, PEG–C(O)–CH2–CH2–S–CH2–), 4.23–4.28 (m, 6H, –CH2–O–C(O)–(CH2)2–S, S–CH2–CH2–CH2–O–C(O)–, dendron –CH2–O–). FT-IR can be seen in the ESI†.
Formation of self-assembled nanoparticles from amphiphilic linear dendritic block-copolymers
The formation of nanoparticles was achieved by dissolving 10 mg of PEG–Gn–PCL in 4 ml of anhydrous acetone. The polymer solution was subsequently slowly added dropwise using a syringe to a 20 ml of PBS buffer under vigorous stirring. The solution was vigorously stirred for 24 h followed by dialysis in 1 l of PBS using a dialysis membrane with 1 kDa MWCO. The dialysis was allowed to proceed for 72 h and with three changes of buffer. The four most hydrophilic materials, i.e. the PEG2k–G0–PCL60, PEG2k–G1–PCL30, PEG5k–G1–PCL60 and PEG5k–G2–PCL30, were further analyzed by DLS and TEM. Materials with PCL:PEG molar mass ratio higher than 3.4 were unable to form micelles as they rapidly precipitated in the buffer solution. Prior to analysis, the micelle solutions were filtered using a 0.45 µm nylon syringe filter in order to remove dust and aggregates.
In order to determine the critical micelle concentration the fluorescent probe technique with pyrene41 was used.
The drug loading/release capacities were evaluated by encapsulation of doxorubicin (DOX). The micelles were loaded using the following procedure: 69 µl of a stock solution, consisting of DOX in CHCl3 (5 mg ml−1) and 3 molar equivalents of Et3Nvs.DOX, were added drop-wise under stirring to a 10 ml vial containing 3 ml of micelle solution (0.38 mg ml−1). The vials were subsequently left open during the night in order to allow the CHCl3 to evaporate. Free DOX was removed by spin filtration using Amicon Ultra 4 centrifugal filters with a MWCO of 3 kDa. UV-Vis spectroscopy was performed on samples diluted 10 times with DMF:H2O 4:1 (ε = 13050 M−1 cm−1)42 to determine loading efficiency, where the loading efficiency is calculated by the quote Amicelle solution/Afree DOX.
Formation of isoporous films from amphiphilic linear dendritic block-copolymers
The ordered honeycomb films were obtained by casting the polymer films under humid atmosphere. 50 µl solution of PEG–Gn–PCL in benzene at a concentration of 1 g l−1 of polymer were cast on a glass substrate (d = 30 mm). The solution was allowed to evaporate at room temperature (20 °C) in a closed humid chamber (90% relative humidity) leading to the formation of a solid white film. The films were analyzed by optical microscopy and SEM.Results and discussion
Synthesis of linear dendritic hybrid block-copolymers using ROP and click chemistry
Non-toxic and biocompatible bis-MPA dendrons7,8 comprising peripheral hydroxyl groups were initially used as macroinitiators for ROP of ε-CL. The library of star branched polymers was originally synthesized using the alkyne functional dendrons (G0–G4) to comprise PCL chain branches with a DP target of either DPPCL = 30 or DPPCL = 60 per arm. The use of dendrons of different generations enabled the synthesis of block-copolymers that expressed similar mass fraction of PCL and molecular weights but that structurally displayed different architectures. For instance, a 1st generation dendron (alk–G1–PCL60) with two PCL polymer chains (DPPCL = 60) has the same molecular weight as a 2nd generation dendron (alk–G2–PCL30) with four PCL chains with DP 30. Furthermore, allyl functional dendrons were used to synthesize additional materials (all–G1–PCL60 and all–G4–PCL30) suitable for the nowadays popular thiol–ene click chemistry. All star branched materials had yields of ≥90% and were characterized with 1H-NMR, SEC, FT-IR and DSC. As seen in Table 1, the targeted molecular weights of the synthesized materials were in good accordance to the results from 1H-NMR and had narrow molecular weight distributions according to SEC. The molecular weights from SEC were, however, not in good agreement to the targeted values as conventional calibration using linear poly(styrene) standards was used.| Material | Yield (%)a | M n,theo/g mol−1 | M n,NMR,core b/g mol−1 | M n,NMR,end - group b/g mol−1 | M n,SEC c/g mol−1 | PDIc | PCLtheo (wt%) | T m d/°Cd | ΔHmd/J g−1 | T c d/°C | ΔHc/J g−1d |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Calculated assuming 75% conversion. b Calculated using 1H-NMR. c Determined using conventional calibration on SEC. d Determined using DSC. | |||||||||||
| alk–G0– PCL30 | 92 | 3481 | 4400 | 4200 | 6800 | 1.12 | 98 | 52.8 | 78.7 | 31.8 | 77.7 |
| alk–G0– PCL60 | 90 | 6905 | 6700 | 7600 | 10700 |
1.10 | 99 | 54.4 | 77.4 | 32.7 | 79.2 |
| alk–G1– PCL30 | 90 | 7021 | 7200 | 8100 | 11500 |
1.13 | 98 | 51.3 | 67.3 | 29.7 | 67.8 |
| alk–G1– PCL60 | 92 | 13870 |
12400 |
10500 |
22800 |
1.15 | 99 | 54.4 | 64.3 | 30.8 | 64.7 |
| alk–G2– PCL30 | 98 | 14102 |
16800 |
17900 |
21900 |
1.16 | 97 | 52.5 | 66.6 | 25.6 | 67.3 |
| alk–G2– PCL60 | 95 | 27800 |
28900 |
33700 |
41100 |
1.13 | 99 | 54.8 | 65.4 | 29.8 | 66.4 |
| alk–G3– PCL30 | 93 | 28265 |
32700 |
35300 |
39300 |
1.11 | 97 | 52.3 | 66.0 | 28.5 | 68.0 |
| alk–G3– PCL60 | 85 | 55661 |
48400 |
70300 |
62300 |
1.05 | 98 | 53.7 | 63.5 | 26.7 | 63.5 |
| alk–G4– PCL30 | 98 | 56590 |
58900 |
70100 |
58300 |
1.06 | 97 | 51.7 | 67.5 | 27.2 | 68.0 |
| all–G1– PCL60 | 97 | 13872 |
14700 |
15600 |
23000 |
1.11 | 98 | 53.9 | 73.2 | 30.4 | 73.5 |
| all–G4– PCL30 | 94 | 56592 |
34900 |
60700 |
50900 |
1.05 | 50.9 | 72.3 | 21.4 | 72.4 | |
| PEG2k–N33 | 60 | 2024 | — | — | 2700 | 1.07 | 52.2 | 155.8 | 23.6 | 150.5 | |
| PEG5k–N33 | 71 | 5024 | — | — | 6500 | 1.11 | 58.8 | 161.8 | 33.7 | 158.5 | |
| PEG2k – SH | 90 | 2088 | — | — | 2800 | 1.05 | 51.5 | 148.9 | 29.7 | 148.5 | |
| PEG2k–G0– PCL60 | 85 | 8905 | — | — | 11900 |
1.07 | 77 | 50.8 | 82.6 | 28.5 | 89.0 |
| PEG2k–G1– PCL30 | 84 | 9021 | — | — | 12400 |
1.10 | 76 | 49.6 | 55.0 | 27.0 | 51.3 |
| PEG2k–G1– PCL60 | 92 | 15870 |
— | — | 19700 |
1.20 | 86 | 53.8 | 59.9 | 28.6 | 60.8 |
| PEG5k–G1– PCL60 | 85 | 18870 |
— | — | 19200 |
1.16 | 73 | 54.5 | 74.5 | 29.7 | 71.7 |
| PEG2k–G2– PCL30 | 92 | 16102 |
— | — | 18900 |
1.22 | 85 | 51.8 | 58.1 | 26.7 | 60.4 |
| PEG2k–G2– PCL60 | 89 | 29800 |
— | — | 36400 |
1.12 | 92 | 54.5 | 59.9 | 27.8 | 60.1 |
| PEG5k–G2– PCL30 | 87 | 19102 |
— | — | 19200 |
1.21 | 72 | 50.6 | 54.5 | 23.8 | 53.3 |
| PEG5k–G2– PCL60 | 89 | 32800 |
— | — | 30700 |
1.20 | 84 | 53.8 | 50.1 | 30.9 | 54.7 |
| PEG2k–G3– PCL30 | 95 | 30265 |
— | — | 34500 |
1.10 | 91 | 52.9 | 58.3 | 31.7 | 57.7 |
| PEG2k–G3– PCL60 | 94 | 57661 |
— | — | 53900 |
1.06 | 95 | 53.1 | 62.0 | 24.6 | 62.5 |
| PEG5k–G3– PCL30 | 86 | 33265 |
— | — | 33900 |
1.15 | 82 | 51.4 | 61.6 | 25.6 | 57.3 |
| PEG5k–G3– PCL60 | 87 | 60661 |
— | — | 43300 |
1.17 | 90 | 53.5 | 62.7 | 27.1 | 63.2 |
| PEG2k–G4– PCL30 | 91 | 58590 |
— | — | 50200 |
1.08 | 94 | 51.7 | 64.2 | 24.9 | 65.0 |
| PEG5k–G4– PCL30 | 97 | 61590 |
— | — | 36700 |
1.14 | 89 | 51.4 | 55.4 | 27.9 | 58.3 |
| TE –PEG2k–G1– PCL60 | 78 | 15960 |
23600 |
1.16 | 86 | 52.9 | 59.9 | 29.9 | 60.7 | ||
| TE –PEG2k–G4– PCL30 | 80 | 58680 |
46700 |
1.05 | 93 | 50.5 | 65.0 | 25.3 | 65.8 |
DSC (Table 1) revealed that Tm for the materials were only affected by the length of the arms (higher DP gave a higher Tm) rather than generation. Tc, ΔHm and ΔHc displayed a similar trend as Tm with the addition of a lowering effect going from G0 to G1. These observations agree well with earlier publications.43,44
In order to introduce hydrophilicity to the branched PCL precursors, azide functional PEGs of two molecular weights (2000 and 5000 Da) were coupled to the single alkyne located at the dendron core using the highly efficient CuAAC click chemistry, see e.g.Fig. 1 for the G4 dendron. An excess of 1.5 equivalents of the azide functional PEGs was used in order to ensure complete conversion of the alkynes as monitored by 1H-NMR. For the lower generations (G0–G2) heating at 40 °C for ca. 24–72 h was sufficient to afford complete conversion. For the higher generations prolonged reaction times (4–5 days) and/or higher reaction temperature (60 °C) were required in order to reach complete conversion of the alkynes. The length of the PEG also had an influence on the reaction times necessary to achieve full conversion; the longer PEG (PEG5k–N33) generally required slightly longer reaction times than PEG2k–N33. When complete conversion had been established, the excess of PEG could subsequently be removed by precipitation in MeOH. As an illustrative example of the CuAAC approach, 1H-NMR and SEC curves of PEG2k–G1–PCL60 and its starting materials are shown in Fig. 2 (1H-NMR) and 3 (SEC). As can be seen in 1H-NMR (Fig. 2), the doublet at 4.71–4.72 ppm from methylene group next to the triple bond (g) has completely disappeared and a new peak has appeared at 5.25 ppm. The shift and disappearance of the triplet at 3.39 ppm, corresponding to the methylene group next to the azide (e), to 4.55 ppm show that the azide has reacted and excess PEG has effectively been removed. In addition, a new peak at 7.79 ppm corresponding to the proton in the triazole ring (f) can also be seen. In SEC (Fig. 3) a slight decrease in molecular weight is found comparing the alk–G1–PCL60 and PEG2k–G1–CPL60 which is the case for most of the linear dendritic hybrids. This could be an evidence of degradation or an effect of the change in hydrodynamic volume rather than actual loss of molecular weight. Furthermore, the SEC reveals that the excess PEG has been removed. The completion of the reaction could further be confirmed by FT-IR as the signals at 1062 and 1103 cm−1 from the PEG are clearly seen in the spectra of the linear dendritic hybrids as well as a change of the relations of the peaks at 2800–3000 cm−1. As the products from the CuAAC approach had a slightly green colour due to residual copper which is potentially disadvantageous in biomedical applications, a simple copper removal procedure was developed and employed. The amphiphilic block-copolymers were precipitated in MeOH:H2O (1:1) with 1% EDTA, and the products were obtained as white powders.
 | ||
| Fig. 1 Structure of PEG–G4–PCL. | ||
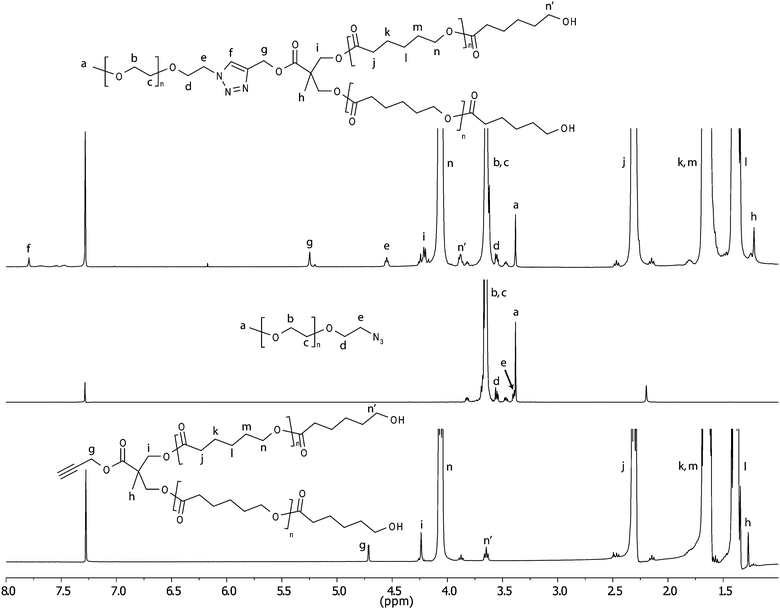 | ||
| Fig. 2 1H-NMR of alk–G1–PCL60, PEG2k–N33 and the formed product PEG2k–G1–PCL60. | ||
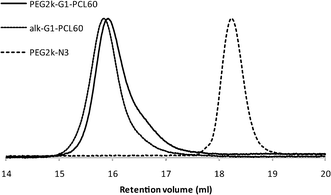 | ||
| Fig. 3 SEC traces of PEG2k–N33, alk–G1–PCL60 and PEG2k–G1–PCL60. | ||
An alternative approach to circumvent the need for additional purification steps of copper catalysts is to employ the bio-friendly thiol–ene click chemistry. The efficacy of the thiol–ene click reaction was initially exploited for the formation of TE–PEG2k–G4–PCL30 and TE–PEG2k–G1–PCL60. In this case, the formation of linear dendritic hybrid block-copolymers required an increased excess of 3 equivalents of PEG–SH, compared to the CuAAC reaction with 1.5 equiv., for the reaction to reach completion. The 1H-NMR of TE–PEG2k–G1–PCL60 and its precursors (all–G1–PCL60 and PEG2k–SH) (Fig. 5) revealed the disappearance of the resonance peak corresponding to the allyl group. The peaks from the allyl (g and f) as well as the peak from the methylene group next to the allyl have all been consumed and new peaks corresponding to the aimed product appeared. Furthermore, the peaks from the PEG2k–SH (a–e) are found supporting the incorporation of the PEG. In SEC (Fig. 4) a slight shift to higher molecular weight is seen as well as evidence of complete removal of the excess PEG. Fig. 4 also shows traces of disulfide coupling of the PEG2k–SH which can be related to the larger excess required for the thiol–ene compared to the CuAAC click reaction.
 | ||
| Fig. 4 SEC traces of PEG2k–SH, all–G1–PCL60 and TE–PEG2k–G1–PCL60. | ||
 | ||
| Fig. 5 1H-NMR of all–G1–PCL60, PEG2k–SH and the formed product TE–PEG2k–G1–PCL60. | ||
Formation of nanoparticles from amphiphilic linear dendritic hybrid block-copolymers and their drug loading capacity
The evaluation of self-assembling properties to micelles for these complex linear dendritic hybrid block-copolymers was found to be limited to structures possessing highest hydophilicity i.e. materials with the lowest PCL:PEG ratio. Consequently, micelles based on PEG2k–G0–PCL60, PEG2k–G1–PCL30, PEG5k–G1–PCL60 and PEG5k–G2–PCL30 were successfully prepared, whereas materials with higher PCL:PEG ratios precipitated upon preparation. The CMCs of the different synthesized materials were determined using the fluorescent probe technique using pyrene as hydrophobic probe (Table 2). Although PEG2k–G0–PCL60 and PEG2k–G1–PCL30 have the similar molecular weight, the influence of having a dendritic wedge is profoundly affecting the CMC for the micelle formation. In fact, PEG2k–G0–PCL60 with no dendritic connector has a CMC of 51 µg ml−1 while PEG2k–G1–PCL30 with a G1 dendron has a CMC of 11 µg ml−1. The same trend can also be seen comparing PEG5k–G1–PCL60 and PEG5k–G2–PCL30 with CMCs of 35 µg ml−1 and 19 µg ml−1 respectively. This effect of lowering the CMC while maintaining the same molecular weight is intriguing and most likely a reflection to which branches in amphiphilic block-copolymers favour the formation of hydrophobic core–shell morphology, compared to linear counterparts. The micelles were further characterised by DLS (Table 2) which showed that increased generation and therefore branches result in larger micelles. This could be seen as an evidence of that a linear dendritic material of a higher generation with shorter arms cannot pack themselves as compact as the one of a lower generation with longer arms. Interestingly, both PEG2k–G1–PCL30 and PEG5k–G1–PCL60 have about the same micelle size according to DLS. This indicates that the architecture rather than the molecular weight is the dominating factor.
| Material | Ratio PCL/PEGtheo | CMCa/µg ml−1 | d DLS,Int . b/nm | d DLS,Vol b/nm | d DLS,Num . b/nm | PDIb | Loading eff. (wt%) |
|---|---|---|---|---|---|---|---|
| a Determined by the fluorescent probe technique. b Determined by DLS at a concentration of 0.34 mg ml−1. | |||||||
| PEG2k–G0– PCL60 | 3.4 | 51 | 98 ± 2 | 36 ± 3 | 21 ± 3 | 0.26 ± 0.01 | 18 |
| PEG2k–G1– PCL30 | 3.4 | 11 | 133 ± 2 | 77 ± 3 | 31 ± 3 | 0.17 ± 0.02 | 17 |
| PEG5k–G1– PCL60 | 2.7 | 35 | 113 ± 7 | 50 ± 2 | 33 ± 2 | 0.27 ± 0.01 | 16 |
| PEG5k–G2– PCL30 | 2.7 | 19 | 79 ± 3 | 54 ± 2 | 41 ± 2 | 0.15 ± 0.01 | 7 |
To investigate the micelles' ability to encapsulate a hydrophobic drug, the potent anti-tumour drug DOX was evaluated (Table 2). It was found that micelles based on G0 and G1 structures had similar degree of encapsulation (16–18 wt%) whereas the G2 only encapsulated about 7 wt%. Again this could be seen as an evidence of a lower degree of packing, i.e. lower crystallinity, of the hydrophobic core due to the increased branching. Initial drug release tests indicate that the nanoparticles can afford a sustained release of doxorubicin; however, a more thorough investigation is currently undertaken in our laboratories in order to determine the kinetics of the drug release, intracellular localization, and the therapeutic effect on cancer cells.
Formation of ordered honeycomb membranes
To further exploit the potential of the amphiphilic block-copolymer library, we sought out the possibility for the fabrication of isoporous membranes. In contrast to the self-assembly of micelles, with a PCL:PEG mass ratio of 3.4 or less, honeycomb membrane formation requires a predominant hydrophobic block that is typically noncrystalline. The incorporation of dendritic linkers within complex structures was foreseen to affect the crystallization process during the membrane fabrication. Indeed, the use of block-copolymers comprising no dendritic connector (PEG2k–G0–PCL60) or with lower generation dendrons (PEG2k–G1–PCL30) failed to produce any ordered membranes, independently of solvent or concentration variations. Interestingly, the use of a block-copolymer with a 3rd generation dendron (PEG2k–G3–PCL30) generated ordered structures when drop cast from benzene, (Fig. 6. A) 1 mg of PEG2k–G3–PCL30 in 1 mL of benzene was sufficient to fabricate the ordered structure when cast on a glass substrate in a closed chamber with ca. 90% relative humidity. The evaporation of the benzene induced cooling on the surface of the film resulting in condensation of water droplets from the humid surrounding. Precipitation of PCL predominant amphiphilic polymer occurred at the water/solvent interface, stabilizing the water droplets. After complete evaporation of the solvent, an opaque white film was obtained. Closer investigation of the film revealed an ordered hexagonal porous structure with an average pore size of 3 µm (Fig. 6). The ordered arrangement was maintained over large areas and the membrane was found storage stable >1 month at room temperature. The SEM characterization of the cross-section revealed the 3D structure of the pores with a depth of 1 µm and a wall thickness of 0.2 µm. The high surface area of the films, resulting from their open pore structure, makes honeycomb membranes attractive materials for applications in which enhanced interactions are necessary. In fact, we are currently evaluating the concentration profile of bioactive isoporous membranes during their interaction with human cells and proteins. Currently further studies are performed in our laboratory in order to fully investigate the effect of dendritic branching points in the formation of isoporous membranes.
 | ||
| Fig. 6 Optical microscopy image at two magnifications (A and B) of an isoporous surface fabricated from PEG2k–G3–PCL30. SEM images of the top (C) and cross-section (D) of an isoporous surface fabricated from PEG2k–G3–PCL30. | ||
Conclusions
By using materials well known for their biocompatibility and non-toxicity a library of linear dendritic hybrid block-copolymers with potential use within biomedical applications has successfully been synthesized. This was accomplished using bis-MPA dendrons, having click functional cores and peripheral hydroxyl groups, as efficient macroinitiators for ROP of ε-CL followed by a click reaction to the core moiety. In addition to the well known CuAAC click reaction we have explored the use of thiol–ene click chemistry as means of synthesizing our linear dendritic hybrids. Comparing the two types of click reactions used in this study they both proved successful. Thiol–ene click chemistry has a few advantages compared to CuAAC click chemistry such as shorter reaction times and the lack of heavy metal catalysts. On the other hand, CuAAC click chemistry requires a lower excess of PEG. In order to evaluate some of the potential applications for this type of materials micelles and isoporous membranes were prepared. The generation of the dendron proved to have a profound influence on the characteristics of the micelles enabling tuneable weight fractions of DOX to be encapsulated.Acknowledgements
The authors would like to acknowledge the Swedish research council (VR) for financial support under grants 2006-3617 and 2009-3259. Assistant Professor fellowship from the Knut and Alice Wallenberg Foundation (to AMN), as well as financial support from Åke Wibergs foundation, Karolinska Institutet, The Swedish Medical Nanoscience Center, Carl Bennet AB, and Vinnova, are also gratefully acknowledged. Wubeshet Sahle at the Department of Functional Nano Materials, KTH is acknowledged for his assistance with TEM measurements.Notes and references
- I. Gitsov, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5295–5314 CrossRef CAS.
- I. Gitsov and J. M. J. Frechet, Macromolecules, 1993, 26, 6536–6546 CrossRef CAS.
- C. J. Hawker, K. L. Wooley and J. M. J. Frechet, J. Chem. Soc., Perkin Trans. 1, 1993, 1287–1297 RSC.
- C. Hua, S. M. Peng and C. M. Dong, Macromolecules, 2008, 41, 6686–6695 CrossRef CAS.
- J. Kohn, S. Abramson and R. Langer, in Biomaterials Science—an Introduction to Materials in Medicine, ed. B. D. Ratner, A. S. Hoffman, F. J. Schoen and J. E. Lemons, Elsevier Academic Press, London, UK, 2nd edn, 2004, pp. 115–127 Search PubMed.
- N. A. Peppas, in Biomaterials Science—an Introduction to Materials in Medicine, ed. B. D. Ratner, A. S. Hoffman, F. J. Schoen and J. E. Lemons, Elsevier Academic Press, London, UK, 2nd edn, 2004, pp. 100–107 Search PubMed.
- O. L. P. De Jesus, H. R. Ihre, L. Gagne, J. M. J. Frechet and F. C. Szoka, Bioconjugate Chem., 2002, 13, 453–461 CrossRef CAS.
- E. R. Gillies, E. Dy, J. M. J. Frechet and F. C. Szoka, Mol. Pharmacol., 2005, 2, 129–138 CrossRef CAS.
- H. Ihre, O. L. P. De Jesus and J. M. J. Frechet, J. Am. Chem. Soc., 2001, 123, 5908–5917 CrossRef CAS.
- H. Ihre, A. Hult, J. M. J. Frechet and I. Gitsov, Macromolecules, 1998, 31, 4061–4068 CrossRef CAS.
- H. Ihre, A. Hult and E. Soderlind, J. Am. Chem. Soc., 1996, 118, 6388–6395 CrossRef CAS.
- C. C. Lee, E. R. Gillies, M. E. Fox, S. J. Guillaudeu, J. M. J. Frechet, E. E. Dy and F. C. Szoka, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 16649–16654 CrossRef CAS.
- M. Malkoch, E. Malmstrom and A. Hult, Macromolecules, 2002, 35, 8307–8314 CrossRef CAS.
- H. Claesson, E. Malmstrom, M. Johansson and A. Hult, Polymer, 2002, 43, 3511–3518 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS.
- C. W. Tornoe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef CAS.
- W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2008, 29, 952–981 CrossRef CAS.
- A. Carlmark, C. J. Hawker, A. Hult and M. Malkoch, Chem. Soc. Rev., 2009, 38, 352–362 RSC.
- G. Franc and A. K. Kakkar, Chem.–Eur. J., 2009, 15, 5630–5639 CrossRef CAS.
- P. L. Golas and K. Matyjaszewski, Chem. Soc. Rev., 2010, 39, 1338–1354 RSC.
- J. A. Johnson, M. G. Finn, J. T. Koberstein and N. J. Turro, Macromol. Rapid Commun., 2008, 29, 1052–1072 CrossRef CAS.
- B. Le Drournaguet and K. Velonia, Macromol. Rapid Commun., 2008, 29, 1073–1089 CrossRef.
- P. Lecomte, R. Riva, C. Jerome and R. Jerome, Macromol. Rapid Commun., 2008, 29, 982–997 CrossRef CAS.
- P. Lundberg, C. J. Hawker, A. Hult and M. Malkoch, Macromol. Rapid Commun., 2008, 29, 998–1015 CrossRef CAS.
- M. Meldal, Macromol. Rapid Commun., 2008, 29, 1016–1051 CrossRef CAS.
- B. S. Sumerlin and A. P. Vogt, Macromolecules, 2010, 43, 1–13 CrossRef CAS.
- M. van Dijk, D. T. S. Rijkers, R. M. J. Liskamp, C. F. van Nostrum and W. E. Hennink, Bioconjugate Chem., 2009, 20, 2001–2016 CrossRef CAS.
- R. K. Iha, K. L. Wooley, A. M. Nystrom, D. J. Burke, M. J. Kade and C. J. Hawker, Chem. Rev., 2009, 109, 5620–5686 CrossRef CAS.
- L. M. Campos, K. L. Killops, R. Sakai, J. M. J. Paulusse, D. Damiron, E. Drockenmuller, B. W. Messmore and C. J. Hawker, Macromolecules, 2008, 41, 7063–7070 CrossRef CAS.
- C. E. Hoyle, T. Y. Lee and T. Roper, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5301–5338 CrossRef CAS.
- K. L. Killops, L. M. Campos and C. J. Hawker, J. Am. Chem. Soc., 2008, 130, 5062–5064 CrossRef CAS.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS.
- C. E. Hoyle, A. B. Lowe and C. N. Bowman, Chem. Soc. Rev., 2010, 39, 1355–1387 RSC.
- M. J. Kade, D. J. Burke and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 743–750 CrossRef CAS.
- V. P. Torchilin, J. Controlled Release, 2001, 73, 137–172 CrossRef CAS.
- G. Widawski, M. Rawiso and B. Francois, Nature, 1994, 369, 387–389 CrossRef CAS.
- U. H. F. Bunz, Adv. Mater., 2006, 18, 973–989 CrossRef CAS.
- P. Wu, A. K. Feldman, A. K. Nugent, C. J. Hawker, A. Scheel, B. Voit, J. Pyun, J. M. J. Frechet, K. B. Sharpless and V. V. Fokin, Angew. Chem., Int. Ed., 2004, 43, 3928–3932 CrossRef CAS.
- R. V. Ostaci, D. Damiron, S. Capponi, G. Vignaud, L. Leger, Y. Grohens and E. Drockenmuller, Langmuir, 2008, 24, 2732–2739 CrossRef CAS.
- K. Kalyanasundaram and J. K. Thomas, J. Am. Chem. Soc., 1977, 99, 2039–2044 CrossRef CAS.
- A. M. Nystrom, Z. Q. Xu, J. Q. Xu, S. Taylor, T. Nittis, S. A. Stewart, J. Leonard and K. L. Wooley, Chem. Commun., 2008, 3579–3581 RSC.
- E. Nunez, C. Ferrando, E. Malmstrom, H. Claesson and U. W. Gedde, J. Macromol. Sci., Part B: Phys., 2004, 43, 1143–1160 Search PubMed.
- E. Nunez, C. Ferrando, E. Malmstrom, H. Claesson, P. E. Werner and U. W. Gedde, Polymer, 2004, 45, 5251–5263 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed information regarding the synthesis of all materials as well as IR spectra of all synthesized materials; further analysis of the micelles including CMC plots and TEM pictures. See DOI: 10.1039/c0py00258e |
| ‡ This paper is part of a Polymer Chemistry issue highlighting the work of emerging investigators in the polymer chemistry field. Guest Editors: Rachel O'Reilly and Andrew Dove. |
| This journal is © The Royal Society of Chemistry 2011 |