Thermo- and light-regulated fluorescence resonance energy transfer processes within dually responsive microgels†‡
Jun
Yin
,
Haibo
Hu
,
Yonghao
Wu
and
Shiyong
Liu
*
CAS Key Laboratory of Soft Matter Chemistry, Department of Polymer Science and Engineering, Hefei National Laboratory for Physical Sciences at the Microscale, University of Science and Technology of China, Hefei, Anhui 230026, China. E-mail: sliu@ustc.edu.cn; Fax: +86-551-360-7348; Tel: +86-551-360-7348
First published on 28th September 2010
Abstract
We report on the fabrication of thermo- and light-responsive P(NIPAM-DMNA-NBDAE-RhBEA) microgels consisting of N-isopropylacrylamide (NIPAM), photocleavable moieties, 5-(2′-(dimethylamino)ethoxy)-2-nitrobenzyl acrylate (DMNA), fluorescence resonance energy transfer (FRET) donors, 4-(2-acryloyloxyethylamino)-7-nitro-2,1,3-benzoxadiazole (NBDAE), and rhodamine B-based FRET acceptors (RhBEA) via the free radical emulsion polymerization technique. Thermo-induced collapse and swelling of responsive microgels above and below the lower critical solution temperatures (LCSTs), respectively, can finely tune the spatial proximity between NBDAE and RhBEA dyes, leading to the facile modulation of FRET efficiencies. Upon UV irradiation (pH 8.5), DMNA moieties within microgels undergo photolysis reactions and the formation of sodium carboxylate residues on microgel scaffolds results in elevated LCST. Thus, UV irradiation of microgel dispersions in the intermediate temperature range (between the LCST of original microgels and that of UV-irradiated microgels) can directly lead to the re-swelling of initially collapsed microgels. The incorporation of FRET pairs (NBDAE and RhBEA dyes) allow for the in situ monitoring of thermo- and UV irradiation-induced volume phase transitions (VPTs) of the reported dually responsive microgels. This work represents the first report of thermoresponsive microgels with VPTs tunable by photolabile 2-nitrobenzyl ester moieties.
Introduction
In the past two decades, ever-increasing attention has been paid to the field of responsive polymers,1–6 which can exhibit reversible or irreversible changes in physical properties and/or chemical structures to an external stimulus such as pH,7–15 temperature,8,9,12,16–22 ionic strength,10 light irradiation,18,22–32 mechanical forces,33,34 electric and magnetic fields,35,36 specific analytes,37 external additives (ions, bioactive molecules, etc.) or a combination of them.38,39 Of these stimuli, light irradiation is quite attractive due to its easy operation, non-invasiveness, low cost, fast responsiveness, and good penetration depth, as compared to conventional stimuli such as pH, temperature, ionic strengths, and other additives. Photo-triggered reactions have been frequently employed in specific applications such as surface patterning,40 structural stabilization41–44 and transformation of molecular assemblies,25,45 caged fluorescence for imaging,46,47 targeted and triggered release of drugs,24,48,49 proteins,50–52 and DNA.53–56Representing a special type of soft matter entities, responsive polymeric hydrogels or microgels have aroused tremendous interest recently due to their applications in controlled drug delivery, tissue engineering, and sensors.57–59 The combination of light-responsiveness with hydrogels or microgels is highly desirable in practical circumstances. Original examples in this aspect were reported by Irie and Kunwatchakun60,61 and Tanaka and co-workers,62 and they covalently attached photochromic triphenylmethane leuco derivatives, which can dissociate into ion pairs under ultraviolet (UV) irradiation, into polyacrylamide or poly(N-isopropylacrylamide) (PNIPAM) hydrogels. In the latter case, volume phase transitions (VPTs) of thermoresponsive PNIPAM hydrogels can be directly induced by UV irradiation within a suitable temperature range. Bradley et al.63 reported that protonated poly(2-vinylpyridine) microgels collapse in the presence of a 4-hexylphenylazosulfonate surfactant, which possesses photo-cleavable ionic head group, and can re-swell upon UV irradiation. As reported by Suzuki and Tanaka64 and later by Nayak and Lyon,25 PNIPAM gels or microgels covalently embedded with strong visible light-adsorbing moieties (copper chlorophyllin, 488 nm absorption; malachite green, 633 nm absorption) can self-heat under laser irradiation and lead to VPT processes.
Recently, photolabile 2-nitrobenzyl ester moieties have also attracted considerable attention.46,65–72 They possess long-term stability under ambient light, but can be efficiently cleaved under UV irradiation. Previously, the photocleavable properties of the 2-nitrobenzyl ester functionality have been utilized for light-triggered release of model drugs from mesoporous silica particles23,37 and block copolymer micelles,46,47,73 controlled release of amine species for the activation of cellular signaling,74 and targeted release of DNA from DNA/functionalized gold nanoparticle complexes53 and DNA/dendron complexes.75 In a recent example, Kasko and co-workers57 reported poly(ethylene glycol)-based degradable hydrogels crosslinked with 2-nitrobenzyl ester-containing difunctional monomers. The obtained hydrogels can act as excellent cell culture scaffolds with photo-tunable physical and chemical properties.
In the above examples concerning photo-responsive microgels or hydrogels, light induced VPT processes or gel degradation are typically monitored by changes in macroscopic dimensions (swelling or shrinkage of gels) or microscopic dimensions (microgel sizes as determined by light scattering techniques). It would be highly desirable to employ alternate techniques, such as colorimetric or fluorometric methods. Previously, Lyon and co-workers76,77 covalently incorporated fluorescence resonance energy transfer (FRET) donor and acceptor dyes into thermoresponsive PNIPAM-based microgels or core-shell nanoparticles and investigated the thermo-induced VPT process.
In this work, we employed the FRET process to monitor thermo- and light-dually regulated phase transitions of 2-nitrobenzyl ester-containing PNIPAM microgels. P(NIPAM-DMNA-NBDAE-RhBEA) microgels consisting of N-isopropylacrylamide (NIPAM), photocleavable moieties, 5-(2′-(dimethylamino)ethoxy)-2-nitrobenzyl acrylate (DMNA), FRET donors, 4-(2-acryloyl-oxyethylamino)-7-nitro-2,1,3-benzoxadiazole (NBDAE), and rhodamine B-based FRET acceptors (RhBEA) were synthesized via the free radical emulsion polymerization technique (Scheme 1a). Upon UV irradiation at pH 8.5, DMNA moieties within microgels undergo photolysis reactions and the formation of sodium carboxylate residues on microgel scaffolds results in elevated LCST. Thus, UV irradiation of microgel dispersions at the intermediate temperature range (between the LCST of original microgels and that of UV-irradiated microgels) can directly lead to the re-swelling of initially collapsed microgels. The incorporation of FRET pairs (NBDAE and RhBEA dyes) allow for the in situ monitoring of thermo- and UV irradiation-induced volume phase transitions (VPTs) of the reported dually responsive microgels (Scheme 1b). To the best of our knowledge, this work represents the first report of thermoresponsive microgels with VPTs tunable by photolabile 2-nitrobenzyl ester moieties.
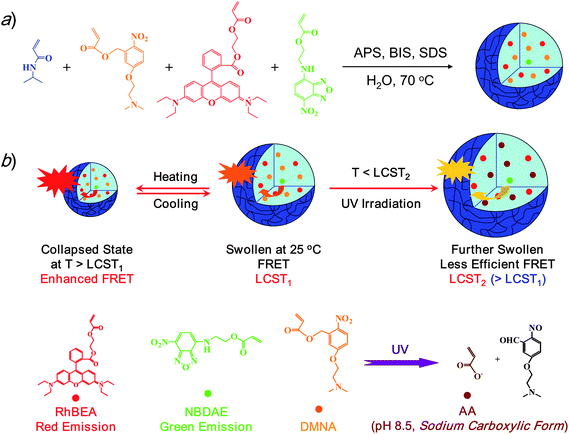 | ||
| Scheme 1 (a) Synthetic schemes employed for the preparation of thermo- and photo-responsive P(NIPAM-DMNA-NBDAE-RhBEA) microgels via emulsion polymerization. (b) A schematic illustration of tuning the efficiency of FRET processes within P(NIPAM-DMNA-NBDAE-RhBEA) microgels by temperature, UV irradiation, or a combination of them. | ||
Experimental
Materials
N-Isopropylacrylamide (NIPAM, 97%, Tokyo Kasei Kagyo Co.) was purified by recrystallization from a mixture of benzene and n-hexane (1/3, v/v). 2-Hydroxyethyl acrylate (HEA, 96%, Tokyo Kasei Kagyo Co.) was purified by distillation under reduced pressure. 3-Hydroxybenzaldehyde (98.5%) and 5-hydroxy-2-nitrobenzaldehyde (1, 98%) were purchased from Aldrich and used as received. Oxalyl chloride (95%), dicyclohexylcarbodiimide (DCC, 99%), triethylamine (TEA), 4-dimethylaminopyridine (DMAP, 99%), rhodamine B (99%), and sodium borohydride (96%) were purchased from Shanghai Chemical Reagent Co. and used as received. 2-(Dimethylamino)ethyl chloride hydrochloride (87%) was recrystallized from ethanol. Acryloyl chloride (Sinopharm Chemical Reagent Co.) was distilled just prior to use. Ammonium persulfate (APS) and N,N′-methylene-bisacrylamide (BIS) were recrystallized from methanol and ethanol, respectively, and stored at −20 °C prior to use. Nonionic surfactant, polyoxyethylene sorbitan monolaurate (Tween 20), was purchased from Amersco and used as received. Methanol, tetrahydrofuran (THF), ethyl acetate (EtOAc), methylene chloride (CH2Cl2), and all other chemicals were purchased from Sinopharm Chemical Reagent Co. Ltd. and used as received. Water was deionized with a Milli-Q SP reagent water system (Millipore) to a specific resistivity of 18.4 MΩcm. 4-(2-Acryloyloxyethylamino)-7-nitro-2,1,3-benzoxadiazole (NBDAE) dye was prepared according to literature procedures.78Synthetic schemes employed for the preparation of photolabile monomer, 5-(2′-(dimethylamino)ethoxy)-2-nitrobenzyl acrylate (DMNA, 4), rhodamine B-based fluorescent monomer (RhBEA, 5), and dually responsive P(NIPAM-DMNA-NBDAE-RhBEA) microgels were shown in Schemes 1 and 2.
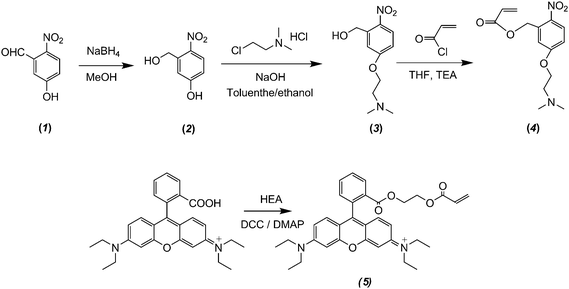 | ||
| Scheme 2 Synthetic routes employed for the preparation of 5-(2′-(dimethylamino)ethoxy)-2-nitrobenzyl acrylate monomer (DMNA, 4) and rhodamine B-based fluorescent monomer (RhBEA, 5). | ||
Synthesis of 5-hydroxy-2-nitrobenzyl alcohol (2)68
A 500 mL round-bottom flask was charged with 1 (20.0 g, 0.12 mol) and methanol (300 mL). After cooling to 0 °C in an ice-water bath, sodium borohydride (9.07 g, 0.24 mol) was slowly added and the resulting solution was stirred at 0 °C. After the reaction was complete, as revealed by thin-layer chromatography (TLC), the reaction mixture was cautiously quenched by the addition of 10% aqueous HCl and the solution pH was adjusted to ∼2, this was followed by extraction with EtOAc (50 mL × 3). The combined organic layers were washed with brine, dried over anhydrous MgSO4, filtered, and then concentrated on rotary evaporator. The crude product was purified by silica gel column chromatography using hexanes/ethyl acetate (1/1, v/v) as the eluent, giving 5-hydroxy-2-nitrobenzyl alcohol (2) as a yellowish solid (19.89 g, 98% yield). 1H NMR (DMSO-d6, δ, ppm, TMS; Fig. S1b‡): 10.88 (s, 1H, Ar–OH), 8.04, 7.24 and 6.78 (m, 3H, aromatic protons), 5.5 (s, 1H, –CH2OH), 4.81 (s, 2H, –CH2OH).Synthesis of 5-(2′-(dimethylamino)ethoxy)-2-nitrobenzyl alcohol (3)53
A 500 mL round-bottom flask was charged with 2 (10.0 g, 59.13 mmol), 2-(dimethylamino)ethyl chloride hydrochloride (7.63 g, 60.95 mmol), sodium hydroxide (5.2 g, 130 mmol), toluene (300 mL), and ethanol (60 mL). The reaction mixture was heated to reflux and stirred for 48 h. After cooling to room temperature, the dark solution was poured into 500 mL of water. The organic layer was isolated and the aqueous phase was further extracted with toluene. The combined organic phase was washed successively with saturated aqueous solution of sodium bicarbonate and water, dried over anhydrous sodium sulfate, and then evaporated to dryness. The crude product was purified by silica gel column chromatography using methanol as the eluent to give 3 as a yellowish solid (10.09 g, yield: 71%). 1H NMR (CDCl3, δ, ppm, TMS; Fig. 1a): 8.14, 7.26, and 6.87 (m, 3H, Aromatic protons), 5.01 (s, 2H, –CH2OH), 4.16 (t, 2H, –OCH2CH2N(CH3)2), 3.03 (s, 1H, –CH2OH), 2.76 (t, 2H, –OCH2CH2N(CH3)2), 2.35 (s, 6H, –OCH2CH2N(CH3)2).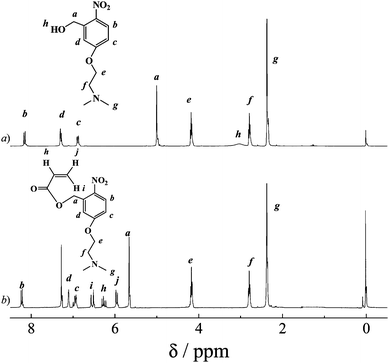 | ||
| Fig. 1 1H NMR spectra recorded in CDCl3 for (a) 5-(2′-(dimethylamino)ethoxy)-2-nitrobenzyl alcohol (3) and (b) 5-(2′-(dimethylamino)ethoxy)-2-nitrobenzyl acrylate monomer (DMNA, 4). | ||
Synthesis of 5-(2′-(dimethylamino)ethoxy)-2-nitrobenzyl acrylate monomer (DMNA, 4)
A 250 mL round-bottom flask was charged with 3 (10.0 g, 41.62 mmol), TEA (4.86 g, 48.07 mmol), and dry THF (50 mL). After cooling to 0 °C in an ice-water bath, acryloyl chloride (3.96 mL, 43.7 mmol) in 10 mL of THF was added dropwise over 1 h. The reaction mixture was then stirred at room temperature for another 5 h. After removing the insoluble salts by suction filtration, the filtrate was concentrated and further purified by silica gel column chromatography using n-hexane–ethyl acetate (3/1, v/v) as the eluent. After removing all the solvents, 4 (DMNA) was obtained as a yellowish solid (9.8 g, yield: 80%). 1H NMR (CDCl3, δ, ppm, TMS; Fig. 1b): 8.2, 7.1, and 6.93 (m, 3H, Aromatic protons), 6.53 (d, 1H, CH2 = CHCOOCH2–), 6.26 (m, 1H, CH2 = CHCOOCH2–), 5.96 (d, 1H, CH2 = CHCOOCH2–), 5.66 (s, 2H, CH2 = CHCOOCH2–), 4.18 (t, 2H, –OCH2CH2N(CH3)2), 2.76 (t, 2H, –OCH2CH2N(CH3)2), 2.34 (s, 6H, –OCH2CH2N(CH3)2).Synthesis of rhodamine B-based fluorescent monomer (RhBEA, 5)
A 50 mL round-bottom flask was charged with rhodamine B (1.0 g, 2.26 mmol), 2-hydroxyethyl acrylate (0.49 g, 4.22 mmol), dicyclohexylcarbodiimide (0.65 g, 3.15 mmol), 4-dimethylaminopyridine (38 mg, 0.31 mmol), and methylene dichloride (15 mL). The reaction mixture was stirred at room temperature for 24 h. After removing the insoluble salts by suction filtration, the filtrate was concentrated and further purified by silica gel column chromatography using ethyl acetate/isopropanol (5/1, v/v) as the eluent. After removing all the solvents, 5 (RhBEA) was obtained as a fuchsia solid (0.61 g, yield: 50%). 1H NMR (CDCl3, δ, ppm, TMS; Fig. 2): 8.31, 7.85, and 7.76, 7.32, 7.08, 6.92, 6.81 (m, 10H, Aromatic protons), 6.37 (dd, 1H, CH2CHCOOCH2CH2O–), 6.07 (dd, 1H, CH2CHCOOCH2CH2O–), 5.87 (dd, 1H, CH2CHCOOCH2CH2O–), 4.29 (m, 2H, CH2CHCOOCH2CH2O–), 4.16 (m, 2H, CH2CHCOOCH2CH2O–), 3.65 (q, 8H, –N(CH2CH3)2), 1.35 (t, 12H, –N(CH2CH3)2).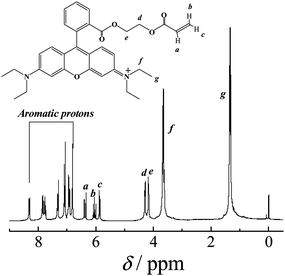 | ||
| Fig. 2 1H NMR spectrum recorded in CDCl3 for rhodamine B-based fluorescent monomer (RhBEA, 5). | ||
Preparation of P(NIPAM-DMNA-NBDAE-RhBEA) microgels
Typical procedures employed for the synthesis of P(NIPAM-DMNA-NBDAE-RhBEA) microgels are as follows. A 250 mL three-necked round-bottom flask equipped with a mechanical stirrer, reflux condenser, and a nitrogen gas inlet was charged with NIPAM (0.80 g, 7.07 mmol), BIS (0.01 g, 64.86 μmol), DMNA monomer (0.20 g, 0.68 mmol), Tween 20 (50 mg, 0.04 mmol), and deionized water (100 mL). After heating to 70 °C and degassing by bubbling with nitrogen for 30 min, APS (0.05 g, 220 μmol) dissolved in 1.0 mL deionized water was injected under mechanical stirring at 400 rpm. NBDAE monomer (10.8 mg, 0.04 mmol) and RhBEA monomer (84 mg, 0.16 mmol) in 2.0 mL ethanol were then added dropwise over ∼20 min. The polymerization was conducted under stirring for 7 h. The dispersion was passed through glass wool in order to remove particulate matter and then dialyzed against deionized water for 5 days. Fresh deionized water was replaced approximately every 6 h. Following similar procedures, the preparation of P(NIPAM-DMNA-NBDAE-RhBEA) microgels with varying DMNA monomer feed ratios (5 and 15 wt% relative to the sum of NIPAM and DMNA) were also prepared (Table 1).| Samples a | DMNA contents (wt%) b | 25 °C | 45 °C | ||
|---|---|---|---|---|---|
| <Rh>/nm c | μ 2/Γ2c | <Rh>/nm c | μ 2/Γ2c | ||
| a All microgel samples had a cross-linking density of 1.0 wt% (relative to the sum of NIPAM and DMNA). b DMNA monomer in the feed recipe relative to the sum of NIPAM and DMNA. c Intensity-average hydrodynamic radius, <Rh>, and polydispersity indices, μ2/Γ2, were determined at pH 8.5 by DLS, and the estimated error associated with <Rh> measurements is ± 2 nm. | |||||
| 1 | 5.0 | 62 | 0.05 | 32 | 0.03 |
| 2 | 15.0 | 67 | 0.07 | 35 | 0.04 |
| 3 | 20.0 | 56 | 0.04 | 30 | 0.02 |
UV-Vis absorption measurements
UV-Vis absorption of P(NIPAM-DMNA-NBDAE-RhBEA) microgel dispersions was measured on a Unico UV/vis 2802PCS spectrophotometer.Temperature-dependent turbidimetry
The optical transmittance of aqueous solutions at a wavelength of 700 nm was acquired on a Unico UV/vis 2802PCS spectrophotometer. A thermostatically controlled couvette was employed and the heating rate was 0.2 °C min−1. The LCST was defined as the temperature corresponding to ∼1% decrease of optical transmittance.Field-emission scanning electron microscope (FE-SEM)
FE-SEM observations were conducted on a high resolution JEOL JSM-6700 field-emission scanning electron microscopy. The samples for SEM observations were prepared by placing 10 μL of microgel solutions on copper grids successively coated with thin films of Formvar and carbon.Dynamic light scattering (DLS)
A commercial spectrometer (ALV/DLS/SLS-5022F) equipped with a multi-tau digital time correlator (ALV5000) and a cylindrical 22 mW UNIPHASE He–Ne laser (λ0 = 632 nm) as the light source was employed for DLS measurements. Scattered light was collected at a fixed angle of 90° for duration of ∼5 min. Distribution averages and particle size distributions were computed using cumulant analysis and CONTIN routines. All data were averaged over three measurements.Fluorescence measurements
Fluorescence spectra were recorded using a RF-5301/PC (Shimadzu) spectrofluorometer. The temperature of the water-jacketed cell holder was controlled by a programmable circulation bath. The slit widths were set at 5 nm for excitation and 5 nm for emission. For all microgel-based FRET experiments, microgel concentrations were fixed at 1.0 × 10−5 g mL−1.Results and discussion
Preparation of P(NIPAM-DMNA-NBDAE-RhBEA) microgels
Typical synthetic routes employed for the preparation of 5-(2′-(dimethylamino)ethoxy)-2-nitrobenzyl acrylate monomer (DMNA, 4) and dually responsive P(NIPAM-DMNA- NBDAE-RhBEA) microgels are shown in Schemes 1 and 2, respectively. The reaction of 5-hydroxy-2-nitrobenzaldehyde (1) with sodium borohydride leads to 5-hydroxy-2- nitrobenzyl alcohol (2) with a yield of 98%. Its chemical structure was confirmed by 1H NMR analysis (Fig. S1‡): compared to the 1H NMR spectrum of 1, the complete disappearance of characteristic peak at ∼10.26 ppm (aldehyde proton) and two newly present resonance peaks at 4.81 and 5.48 ppm in the 1H NMR spectrum of 2 indicates the successful preparation of 2. This was followed by the nucleophilic substitution reaction of 2-(dimethylamino)ethyl chloride hydrochloride with 2 in the presence of NaOH. In the final step, the target monomer, 5-(2′-(dimethylamino)ethoxy)-2-nitrobenzyl acrylate (DMNA, 4), was obtained by the esterification reaction of 3 with acryloyl chloride. The chemical structures of 3 and 4 were both verified by 1H NMR analysis (Fig. 1). We aim to enhance the hydrophilicity of 2-nitrobenzyl ester-based monomer via the incorporation of tertiary amine residues, which remain non-protonated at ∼pH 8.5 (i.e., the experimental pH condition), but transforms into sodium carboxylate moieties upon UV irradiation. The generation of negative charges on the gel scaffolds can elevate the LCST of thermoresponsive PNIPAM-based microgels.P(NIPAM-DMNA-NBDAE-RhBEA) microgels were synthesized via free radical emulsion copolymerization of NIPAM, photolabile monomer (DMNA), and FRET donor and acceptor-based monomers (NBDAE and RhBEA monomers) in the presence of BIS and Tween 20 at around neutral pH and 70 °C. All microgel samples possess a feed cross-linking density of 1.0 wt%, whereas the DMNA monomer feed contents (relative to the sum of NIPAM and DMNA) varied from 5.0 wt% to 20.0 wt%. As-synthesized microgels were characterized by DLS, and the obtained intensity-average hydrodynamic radius, <Rh>, and polydispersity indices, μ2/Γ2, are summarized in Table 1. The SEM images shown in Fig. 3 reveal the presence of near-monodisperse and spherical nanoparticles with an average diameter of ∼100 nm for all three microgel samples (DMNA feed contents are 5.0 wt%, 15.0 wt%, and 20.0 wt%, respectively), which are in general agreement with those obtained by DLS measurements (Table 1).
 | ||
| Fig. 3 Typical SEM images obtained by drying aqueous dispersions of thermo- and photo-responsive P(NIPAM-DMNA-NBDAE-RhBEA) microgels prepared at varying DMNA feed contents: (a) 5.0 wt%, (b) 15.0 wt%, and (c) 20.0 wt%. Scale bars represent 1 μm in all cases. | ||
Thermo- and light-regulated volume phase transitions of P(NIPAM-DMNA-NBDAE-RhBEA) microgels
Previously, Kim and co-workers23 fabricated a novel type of cyclodextrin (CD)-capped mesoporous silica nano-containers exhibiting photo-triggered release characteristics due to the presence of 2-nitrobenzyl ester moieties, which initially act as covalent links between the capping agent (CD) and the nanopore outlets. The photocleavage of 2-nitrobenzyl moieties was accompanied with the decrease and increase of UV absorbance at 282 nm and at 344 nm, respectively. In the current work, the aqueous dispersion of P(NIPAM-DMNA-NBDAE-RhBEA) microgels was subjected to irradiation with UV light (365 nm) for ∼30 min. UV-Vis spectra of microgel dispersions obtained before and after UV irradiation (pH 8.5, [DMNA] = 20 wt%; Fig. 4) clearly revealed that UV absorbance at 284 nm decreased, accompanied with the increase of absorbance at ∼318 nm. This suggested the photo-triggered cleavage of 2-nitrobenzyl ester moieties and the generation of 2-nitrosobenzaldehyde derivatives and sodium carboxylate residues. Thus, initially non-charged microgels will bear net negative charges upon UV irradiation.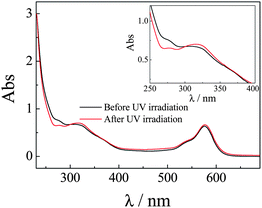 | ||
| Fig. 4 UV-vis spectra recorded for P(NIPAM-DMNA-NBDAE-RhBEA) microgel dispersions (pH 8.5, 1.0 × 10−4 g mL−1, 25 °C; microgels were prepared with a DMNA feed ratio of 20.0 wt%) before and after UV irradiation (365 nm) for 30 min. | ||
PNIPAM homopolymers and gels are well-known to undergo phase transitions upon heating to above the LCST at ∼32 °C. The incorporation of hydrophilic or hydrophobic moieties into PNIPAM can appreciably increase or decrease the LCSTs, respectively. Previously, Irie and Kunwatchakun60,61 and Tanaka and co-workers62 covalently attached triphenylmethane leuco derivatives into PNIPAM gels. Under UV irradiation, the generation of positive charges from triphenylmethane leuco derivatives can elevate the LCST of gels and lead to photo-triggered phase transitions.
We then employed temperature-dependent optical transmittance to determine the changes of LCSTs of P(NIPAM-DMNA-NBDAE-RhBEA) microgels before and after UV irradiation (Fig. 5). For three types of microgel dispersions with varying DMNA contents (5.0 wt%, 15.0 wt%, and 20.0 wt%) at pH 8.5 before irradiation, they possess LCSTs of 29 °C, 30 °C, and 30 °C, respectively. Note that LCSTs were defined as the temperature corresponding to ∼1% decrease in optical transmittance. We found that upon UV irradiation for 30 min, LCSTs considerably shifted to 34 °C and 39 °C for microgel samples prepared at DMNA feed contents of 15.0 wt% and 20.0 wt%, respectively; whereas at a DMNA feed content of 5.0 wt%, the LCST only slightly shifted to 29.5 °C after UV irradiation. At pH 8.5 upon UV irradiation, neutral DMNA moieties dissociate into 2-nitrosobenzaldehyde small molecule derivatives and negative charged sodium carboxylate residues attached to the microgel networks. Increased hydrophilicity and repulsion between negatively charged species lead to the increase of LCSTs. For microgel samples with a DMNA content of 5.0 wt%, the increase of LCST is quite modest, compared to those with DMNA contents of 15.0 wt% and 20.0 wt%. This indicates that for microgels with low DMNA contents, photo-generated negative charges and increased hydrophilicity cannot exhibit appreciable effects on its phase transition temperatures. In the next section, we focused on the monitoring of thermo- and light-regulated microgel collapse and swelling through the spectrofluorometric technique by utilizing the introduced FRET pairs (NBDAE and RhBEA dyes).
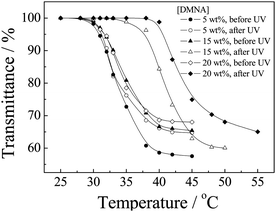 | ||
| Fig. 5 Temperature dependence of normalized optical transmittance at a wavelength of 700 nm obtained for aqueous dispersions of P(NIPAM-DMNA-NBDAE-RhBEA) microgels (pH 8.5, 1.0 × 10−4 g mL−1) prepared at varying DMNA feed contents (5.0 wt%, 15.0 wt%, and 20.0 wt%) before and after UV irradiation (365 nm) for 30 min. LCSTs were defined as the temperature corresponding to ∼1% decrease in optical transmittance. | ||
The phase transitions of thermoresponsive microgels are typically monitored by thermo-induced size changes determined by DLS or by temperature-dependent optical transmittance. As shown previously by Lyon and co-workers,76,77 the introduction of FRET processes within thermoresponsive microgels can lead to more sensitive monitoring of VPTs. They synthesized fluorescently labeled core-shell PNIPAM microgels covalently incorporated with FRET donors (phenanthrene or Cy5) and FRET acceptors (anthracene or Cy5.5) to probe thermo-induced the gel swelling and collapsing behavior. The considerable changes in microgel dimensions during phase transition can apparently lead to changes in spatial distances between FRET donors and acceptors embedded within microgels. For P(NIPAM-DMNA-NBDAE-RhBEA) microgels with a DMNA feed ratio of 20.0 wt%, the <Rh> decreased from 56 nm at 25 °C to 30 nm at 45 °C, i.e., ∼6.5 times decrease in the hydrodynamic volume.
P(NIPAM-DMNA-NBDAE-RhBEA) microgels contain NBDAE and RhBEA dyes, and they can act as FRET donor and acceptors, respectively. For microgel dispersion before UV irradiation, temperature-dependent fluorescence spectra were acquired at first and the results are shown in Fig. 6–7. From Fig. 6a, we can apparently observe two emission peaks at around 527 nm and 588 nm, which can be ascribed to emissions of NBDAE and RhBEA dyes, respectively. In the temperature range of 25–55 °C, the decrease and increase of emission intensities at 527 nm and 588 nm, respectively, are clearly evident. Fig. 7a plots the changes in fluorescence intensity ratios, F588/F527, as a function of temperatures. For the microgel sample (20.0 wt% DMNA content) before UV irradiation, we can discern an inflection point at ∼30 °C, which correlates well with the LCST determined by temperature-dependent optical transmittance (Fig. 5). This suggests that thermo-induced volume phase transition of microgels leads to closer proximity between FRET donors and acceptors, i.e., the increase of FRET efficiencies. The use of FRET efficiencies to monitor the VPT of thermoresponsive microgels has been previously established by the Lyon research group.76,77
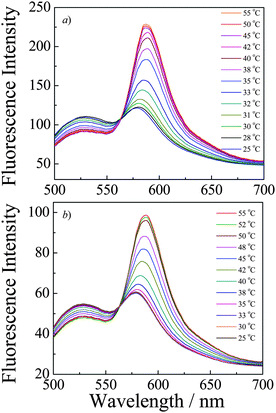 | ||
| Fig. 6 Fluorescence emission spectra (λex = 470 nm; slit widths: Ex. 5 nm, Em. 5 nm) recorded at varying temperatures for P(NIPAM-DMNA-NBDAE-RhBEA) microgel dispersions (pH 8.5, 1.0 × 10−5 g mL−1; microgels were prepared with a DMNA feed ratio of 20.0 wt%) (a) before and (b) after UV irradiation (365 nm) for 30 min. | ||
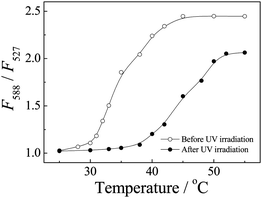 | ||
| Fig. 7 Temperature-dependent fluorescence intensity ratio changes, F588/F527, obtained for P(NIPAM-DMNA-NBDAE-RhBEA) microgel dispersions (pH 8.5, 1.0 × 10−5 g mL−1; microgels were prepared with a DMNA feed ratio of 20.0 wt%) before and after UV irradiation (365 nm) for 30 min. | ||
It is worth noting that in the current system, FRET donors (NBDAE moieties) and acceptors (RhBEA residues) are expected to be randomly distributed within PNIPAM-based microgels, so there exists a distribution of FRET efficiencies between different donor–acceptor species with varying spatial distances. Thus, the obtained FRET efficiencies can only be considered as an averaged value. Moreover, fluorescence emission at ∼588 nm is a sum of contributions from the FRET process and the direct excitation of RhBEA residues. Overall, it is quite difficult to quantify the relative contributions from both aspects.
For P(NIPAM-DMNA-NBDAE-RhBEA) microgels (20.0 wt%) subjected to UV irradiation for 30 min, we also checked the temperature-dependent fluorescence emissions and the results are also plotted in Fig. 7 and 8. The increase of LCST is clearly evident from temperature-dependent fluorescence intensity ratio changes, F588/F527, as shown in Fig. 8b. F588/F527 only exhibit abrupt changes above ∼38 °C, which again correlates very well with the LCST of UV irradiated microgels determined by temperature-dependent optical transmittance (∼39 °C, Fig. 5). As shown in Fig. S2–S3,‡ similar thermo-induced changes of fluorescence intensity ratios have also been observed for microgels with 15.0 wt% of DMNA before and after UV irradiation. This indicates that FRET efficiencies of thermoresponsive microgels can be facilely employed to characterize the phase transitions temperature and its light-regulated shift.
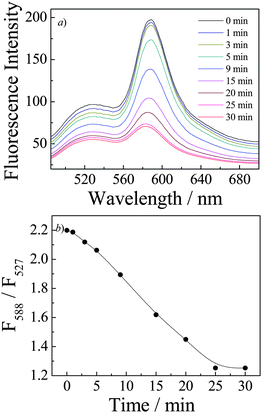 | ||
| Fig. 8 (a) Time evolution of fluorescence emission spectra (λex = 470 nm; slit widths: Ex. 5 nm, Em. 5 nm) and (b) time dependence of fluorescence intensity ratio changes, F588/F527, recorded for P(NIPAM-DMNA-NBDAE-RhBEA) microgel dispersions (pH 8.5, 1.0 × 10−5 g mL−1; microgels were prepared with a DMNA feed ratio of 20.0 wt%) upon UV irradiation (365 nm). | ||
We have now established that UV irradiation of P(NIPAM-DMNA-NBDAE-RhBEA) microgels (20.0 wt% DMNA) can lead to a shift of LCST from ∼30 °C to ∼39 °C (Fig. 5–7). If UV irradiation was conducted at ∼38 °C, i.e., microgels were initially in the collapsed state, we can expect that microgel re-swelling will occur due to the increase of LCST of UV irradiated microgels. The time evolution of fluorescence spectra and fluorescence intensity ratio changes during UV irradiation were then monitored (Fig. 8). We can tell from Fig. 8b the gradual decrease of F588/F527 upon UV irradiation of microgel dispersion. This clearly indicates UV-triggered phase transition of microgels. Again, the changes of FRET efficiencies can be employed to in situ monitor the phase transition process. Photo-induced microgel re-swelling was also confirmed by DLS measurements (Fig. S4‡). For P(NIPAM-DMNA-NBDAE-RhBEA) microgels prepared at DNMA feed content of 20.0 wt%, its dispersion exhibited <Rh> values of 56 nm and 34 nm at 25 °C and 38 °C, respectively. Upon UV irradiation at 38 °C for 30 min, <Rh> considerably increased to ∼60 nm, i.e., prominent photo-induced microgel swelling was observed. It is reasonable that the size dimension of UV-irradiated microgels at 38 °C (∼60 nm) is slightly larger than that of un-irradiated samples at 25 °C (56 nm). UV-irradiated microgels, electrostatic repulsions between UV-generated negative charges within microgels led to their further swelling compared to that of almost neutral microgels.
Fluorescent labeling of P(NIPAM-DMNA-NBDAE-RhBEA) microgels with FRET donors and acceptors also allow for the visual inspection of thermo-induced phase transition and UV-light triggered phase transitions. As shown in Fig. 9, upon heating from 25 °C to 38 °C, the collapse of microgels leads to enhanced FRET efficiency due to closer proximity of NBDAE and RhBEA dyes. Under UV light, we can clearly discern the orange-to-red transition upon heating the microgel dispersion. On the other hand, at a fixed temperature of 38 °C, UV irradiation for 30 min leads to the red-to-orange transition, which is associated with the light-triggered phase transition of microgels (Fig. 9). Due to that the photocleavage of 2-nitrobenzyl ester moieties is irreversible, the above light-triggered phase transition of microgels is also irreversible. However, we successfully established that the FRET processes occurred within responsive microgels can be utilized for monitoring the UV-triggered phase transition processes.
 | ||
| Fig. 9 Photographs immediately recorded under a 365 nm UV Lamp for P(NIPAM-DMNA-NBDAE-RhBEA) microgel dispersions (pH 8.5, 1.0 × 10−4 g mL−1; microgels were prepared with a DMNA feed ratio of 20.0 wt%) under varying conditions: (a) 25 °C; (b) 38 °C; (c) 38 °C and after UV irradiation (365 nm) for 30 min. | ||
Conclusions
In summary, we fabricated thermo- and light-responsive P(NIPAM-DMNA-NBDAE-RhBEA) microgels consisting of photocleavable moieties, DMNA, fluorescence resonance energy transfer (FRET) donors, NBDAE, and rhodamine B-based FRET acceptors, RhBEA, via the free radical emulsion polymerization technique. FRET efficiencies between NBDAE and RhBEA moieties can be employed to monitor the thermo-induced microgel collapse and swelling. Moreover, UV irradiation of P(NIPAM-DMNA-NBDAE-RhBEA) microgels can considerably elevate the LCSTs due to the generation of negatively charged sodium carboxylate species within microgels, as evidenced from temperature-dependent changes in optical transmittance and FRET efficiencies. At temperatures located between the LCSTs of non-irradiated and UV-irradiated microgel dispersion, UV-triggered phase transitions can be achieved and the transition process can also be monitored by changes in FRET efficiencies.Acknowledgements
The financial support of National Natural Scientific Foundation of China (NNSFC) Projects (20874092 and 51033005) and Specialized Research Fund for the Doctoral Program of Higher Education (SRFDP) is gratefully acknowledged.References
- A. Nelson, Nat. Mater., 2008, 7, 523–525 CrossRef CAS.
- C. D. Vo, J. Rosselgong, S. P. Armes and N. Tirelli, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2032–2043 CrossRef CAS.
- J. P. Wang, D. J. Gan, L. A. Lyon and M. A. El-Sayed, J. Am. Chem. Soc., 2001, 123, 11284–11289 CrossRef CAS.
- D. C. Wu, Y. Liu and C. B. He, Macromolecules, 2008, 41, 18–20 CrossRef CAS.
- N. T. Zaman, Y. Y. Yang and J. Y. Ying, Nano Today, 2010, 5, 9–14 CrossRef CAS.
- B. Mattiasson, A. Kumar, A. E. Ivanov and I. Y. Galaev, Nat. Protoc., 2007, 2, 213–220 Search PubMed.
- W. B. Liechty, R. J. Chen, F. Farzaneh, M. Tavassoli and N. K. H. Slater, Adv. Mater., 2009, 21, 3910 CrossRef CAS.
- D. Schmaljohann, Adv. Drug Delivery Rev., 2006, 58, 1655–1670 CrossRef CAS.
- A. S. Carreira, F. A. M. M. Goncalves, P. V. Mendonca, M. H. Gil and J. F. J. Coelho, Carbohydr. Polym., 2010, 80, 618–630 CrossRef CAS.
- J. Huang, X. B. Hu, W. X. Zhang, Y. H. Zhang and G. T. Li, Colloid Polym. Sci., 2008, 286, 113–118 CrossRef CAS.
- V. C. Ibekwe, H. M. Fadda, G. E. Parsons and A. W. Basit, Int. J. Pharm., 2006, 308, 52–60 CrossRef CAS.
- K. Kurata and A. Dobashi, J. Macromol. Sci., Pure Appl. Chem., 2004, A41, 143–164 CrossRef CAS.
- Y. Oda, S. Kanaoka and S. Aoshima, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1207–1213 CrossRef CAS.
- L. A. Connal, Q. Li, J. F. Quinn, E. Tjipto, F. Caruso and G. G. Qiao, Macromolecules, 2008, 41, 2620–2626 CrossRef CAS.
- S. Dai, P. Ravi and K. C. Tam, Soft Matter, 2008, 4, 435–449 RSC.
- R. Pelton, Adv. Colloid Interface Sci., 2000, 85, 1–33 CrossRef CAS.
- S. V. Ghugare, P. Mozetic and G. Paradossi, Biomacromolecules, 2009, 10, 1589–1596 CrossRef CAS.
- C. H. Luo, F. Zuo, Z. H. Zheng, X. B. Ding and Y. X. Peng, J. Macromol. Sci., Part A: Pure Appl. Chem., 2008, 45, 364–371 Search PubMed.
- K. Iwai, Y. Matsumura, S. Uchiyama and A. P. de Silva, J. Mater. Chem., 2005, 15, 2796–2800 RSC.
- V. V. Khutoryanskiy and G. A. Mun, J. Pharm. Pharmacol., 2008, 60, A12–A12.
- M. Kano and E. Kokufuta, Langmuir, 2009, 25, 8649–8655 CrossRef CAS.
- F. D. Jochum and P. Theato, Macromolecules, 2009, 42, 5941–5945 CrossRef CAS.
- C. Park, K. Lee and C. Kim, Angew. Chem., Int. Ed., 2009, 48, 1275–1278 CrossRef CAS.
- M. S. Kim and S. L. Diamond, Bioorg. Med. Chem. Lett., 2006, 16, 4007–4010 CrossRef CAS.
- S. Nayak and L. A. Lyon, Chem. Mater., 2004, 16, 2623–2627 CrossRef CAS.
- M. J. Tucker, J. R. Courter, J. Chen, O. Atasoylu, I. A. B. Smith and R. M. Hochstrasser, Angew. Chem., Int. Ed., 2010, 49, 1–6.
- W. Deng, M. H. Li, X. G. Wang and P. Keller, Liq. Cryst., 2009, 36, 1023–1029 CrossRef CAS.
- F. D. Jochum, L. zur Borg, P. J. Roth and P. Theato, Macromolecules, 2009, 42, 7854–7862 CrossRef CAS.
- M. J. Lee, D. H. Jung and Y. K. Han, Mol. Cryst. Liq. Cryst., 2006, 444, 41–50 CrossRef CAS.
- T. Dvir, M. R. Banghart, B. P. Timko, R. Langer and D. S. Kohane, Nano Lett., 2010, 10, 250–254 CrossRef CAS.
- G. D. Jaycox, Polymer, 2007, 48, 82–90 CrossRef CAS.
- C. G. Bochet, Tetrahedron Lett., 2000, 41, 6341–6346 CrossRef CAS.
- M. Stieger and W. Richtering, Macromolecules, 2003, 36, 8811–8818 CrossRef CAS.
- Y. Sagara and T. Kato, Nat. Chem., 2009, 1, 605–610 Search PubMed.
- C. S. Brazel, Pharm. Res., 2009, 26, 644–656 CrossRef CAS.
- M. Irie, Macromolecules, 1986, 19, 2890–2892 CrossRef CAS.
- C. Park, H. Kim, S. Kim and C. Kim, J. Am. Chem. Soc., 2009, 131, 16614 CrossRef CAS.
- J. P. Magnusson, A. Khan, G. Pasparakis, A. O. Saeed, W. X. Wang and C. Alexander, J. Am. Chem. Soc., 2008, 130, 10852–10853 CrossRef CAS.
- M. Bradley, J. Ramos and B. Vincent, Langmuir, 2005, 21, 1209–1215 CrossRef CAS.
- D. Ryan, B. A. Parviz, V. Linder, V. Semetey, S. K. Sia, J. Su, M. Mrksich and G. M. Whitesides, Langmuir, 2004, 20, 9080–9088 CrossRef CAS.
- A. Housni, M. Ahmed, S. Y. Liu and R. Narain, J. Phys. Chem. C, 2008, 112, 12282–12290 CrossRef CAS.
- J. Q. Jiang, B. Qi, M. Lepage and Y. Zhao, Macromolecules, 2007, 40, 790–792 CrossRef CAS.
- X. Z. Jiang, S. Z. Luo, S. P. Armes, W. F. Shi and S. Y. Liu, Macromolecules, 2006, 39, 5987–5994 CrossRef CAS.
- Q. Jin, X. S. Liu, G. Y. Liu and J. Ji, Polymer, 2010, 51, 1311–1319 CrossRef CAS.
- J. Ruchmann, S. Fouilloux and C. Tribet, Soft Matter, 2008, 4, 2098–2108 RSC.
- M. Lepage, J. Q. Jiang, J. Babin, B. Qi, L. Tremblay and Y. Zhao, Phys. Med. Biol., 2007, 52, N249–N255 CrossRef CAS.
- J. Q. Jiang, X. Tong, D. Morris and Y. Zhao, Macromolecules, 2006, 39, 4633–4640 CrossRef CAS.
- S. S. Agasti, A. Chompoosor, C. C. You, P. Ghosh, C. K. Kim and V. M. Rotello, J. Am. Chem. Soc., 2009, 131, 5728–5729 CrossRef CAS.
- J. L. Vivero-Escoto, I. I. Slowing, C. W. Wu and V. S. Y. Lin, J. Am. Chem. Soc., 2009, 131, 3462 CrossRef CAS.
- M. M. Felix, H. Umakoshi, T. Shimanouchi, M. Yoshimoto and R. Kuboi, Biochem. Eng. J., 2002, 12, 7–19 CrossRef CAS.
- M. S. Kim, J. Gruneich, H. Y. Jing and S. L. Diamond, J. Mater. Chem., 2010, 20, 3396–3403 RSC.
- F. S. Du, Y. Wang, R. Zhang and Z. C. Li, Soft Matter, 2010, 6, 835–848 RSC.
- G. Han, C. C. You, B. J. Kim, R. S. Turingan, N. S. Forbes, C. T. Martin and V. M. Rotello, Angew. Chem., Int. Ed., 2006, 45, 3165–3169 CrossRef CAS.
- M. A. Kostiainen, D. K. Smith and O. Ikkala, Angew. Chem., Int. Ed., 2007, 46, 7600–7604 CrossRef.
- G. H. McGall, A. D. Barone, M. Diggelmann, S. P. A. Fodor, E. Gentalen and N. Ngo, J. Am. Chem. Soc., 1997, 119, 5081–5090 CrossRef CAS.
- A. C. Pease, D. Solas, E. J. Sullivan, M. T. Cronin, C. P. Holmes and S. P. A. Fodor, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 5022–5026.
- A. M. Kloxin, A. M. Kasko, C. N. Salinas and K. S. Anseth, Science, 2009, 324, 59–63 CrossRef CAS.
- J. Yin, X. F. Guan, D. Wang and S. Y. Liu, Langmuir, 2009, 25, 11367–11374 CrossRef CAS.
- T. Liu, J. M. Hu, J. Yin, Y. F. Zhang, C. H. Li and S. Y. Liu, Chem. Mater., 2009, 21, 3439–3446 CrossRef CAS.
- M. Irie and D. Kunwatchakun, Makromol. Chem. Rapid Commun., 1985, 5, 829–832.
- M. Irie and D. Kunwatchakun, Macromolecules, 1986, 9, 2476–2480 CrossRef.
- A. Mamada, T. Tanaka, D. Kungwatchakun and M. Irie, Macromolecules, 1990, 23, 1517–1519 CrossRef CAS.
- M. Bradley, B. Vincent, N. Warren, J. Eastoe and A. Vesperinas, Langmuir, 2006, 22, 101–105 CrossRef CAS.
- A. Suzuki and T. Tanaka, Nature, 1990, 346, 345–347 CrossRef CAS.
- M. Schworer and J. Wirz, Helv. Chim. Acta, 2001, 84, 1441–1458 CrossRef CAS.
- J. Hu, J. Zhang, F. Liu, K. Kittredge, J. K. Whitesell and M. A. Fox, J. Am. Chem. Soc., 2001, 123, 1464–1470 CrossRef CAS.
- Y. V. Il'ichev, M. A. Schworer and J. Wirz, J. Am. Chem. Soc., 2004, 126, 4581–4595 CrossRef CAS.
- M. Kang and B. Moon, Macromolecules, 2009, 42, 455–458 CrossRef CAS.
- D. Y. Wong, D. R. Griffin, J. Reed and A. M. Kasko, Macromolecules, 2010, 43, 2824–2831 CrossRef CAS.
- M. Gaplovsky, Y. V. Il'ichev, Y. Kamdzhilov, S. V. Kombarova, M. Mac, M. A. Schworer and J. Wirz, Photochem. Photobiol. Sci., 2005, 4, 33–42 RSC.
- F. Bley, K. Schaper and H. Gorner, Photochem. Photobiol., 2008, 84, 162–171 CAS.
- K. H. Choi, J. C. Jung, K. S. Kim and J. B. Kim, Polym. Adv. Technol., 2005, 16, 387–392 CrossRef CAS.
- Y. Zhao, J. Mater. Chem., 2009, 19, 4887–4895 RSC.
- J. Nakanishi, H. Nakayama, T. Shimizu, H. Ishida, Y. Kikuchi, K. Yamaguchi and Y. Horiike, J. Am. Chem. Soc., 2009, 131, 3822 CrossRef CAS.
- M. A. Kostiainen, O. Kasyutich, J. J. L. M. Cornelissen and R. J. M. Nolte, Nat. Chem., 2010, 2, 394–399 Search PubMed.
- C. D. Jones, J. G. McGrath and L. A. Lyon, J. Phys. Chem. B, 2004, 108, 12652–12657 CrossRef CAS.
- J. P. Wang, D. J. Gan, L. A. Lyon and M. A. El-Sayed, J. Am. Chem. Soc., 2001, 123, 11284–11289 CrossRef CAS.
- S. Uchiyama, Y. Matsumura, A. P. de Silva and K. Iwai, Anal. Chem., 2003, 75, 5926–5935 CrossRef CAS.
Footnotes |
| † Footnote: This paper is part of a Polymer Chemistry issue highlighting the work of emerging investigators in the polymer chemistry field. Guest Editors: Rachel O'Reilly and Andrew Dove. |
| ‡ Electronic supplementary information (ESI) available: 1H NMR data of 5-hydroxy-2-nitrobenzaldehyde (1) and 5-hydroxy-2-nitrobenzyl alcohol (2), fluorescence characterization data for microgels at varying temperatures, and typical hydrodynamic radius distributions of microgels at varying temperatures before and after UV irradiation. See DOI: 10.1039/c0py00254b |
| This journal is © The Royal Society of Chemistry 2011 |