Organocatalytic synthesis of astaxanthin-containing poly(lactide)s†
Helen
Middleton
,
Sarah
Tempelaar
,
David M.
Haddleton
and
Andrew P.
Dove
*
Department of Chemistry, University of Warwick, Coventry, CV4 7AL, UK. E-mail: a.p.dove@warwick.ac.uk; Tel: +44 (0)24 7652 4107
First published on 20th December 2010
Abstract
The synthesis of astaxanthin-containing poly(lactide)s is reported by the ring-opening polymerization of lactide initiated from residual alcohol groups on astaxanthin using a previously reported thiourea/tertiary amine catalyst. Polymers with molecular weights between 2500 and 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 are obtained with excellent levels of control, astaxanthin incorporation being confirmed by UV/Vis detected GPC, 1H NMR, MALDI-TOF MS and IR spectroscopic analysis. Study of the polymerizations at extended time periods revealed greatly increased levels of transesterification in comparison to polymerizations initiated by 4-pyrene-1-butanol, attributed to increased intramolecular transesterification side reactions.
000 g mol−1 are obtained with excellent levels of control, astaxanthin incorporation being confirmed by UV/Vis detected GPC, 1H NMR, MALDI-TOF MS and IR spectroscopic analysis. Study of the polymerizations at extended time periods revealed greatly increased levels of transesterification in comparison to polymerizations initiated by 4-pyrene-1-butanol, attributed to increased intramolecular transesterification side reactions.
Introduction
The incorporation of bioactive molecules in polymeric systems for in vivo delivery applications is an area of high interest. Several controlled release strategies exist including release from micellar systems1,2 and by erosion of hydrophobic polymers.3,4 Poly(lactide), PLA, is often a key component in either approach, largely a consequence of both its biocompatibility and biodegradability.5–10 Where covalent attachment is preferred to yield greater stability and reduced levels of leaching, both ‘grafting-to’ and ‘grafting-from’ strategies have been successful in the attachment of polymers to a range of bioactive molecules ranging from antibiotics to proteins and enzymes.11PLA is commonly synthesized by ring-opening polymerization, ROP, which in turn is typically initiated from alcohol residues.12–14 As such utilization of a ‘grafting-from’ approach, whereby ROP is initiated from the bioactive molecule, presents an excellent opportunity to obtain high and controllable levels of bioactive molecule incorporation in a degradable polymer matrix.The ROP of LA can be catalyzed by a wide range of species including metal-based complexes,12,15,16 enzymes17,18 and simple organic molecules.13,14,19 While all three classes of compound have their own advantages, organic catalysts are often stable to moisture and oxygen, and are highly active and selective for ROP while negating the requirement for the removal of the often highly toxic heavy metals from the resultant polymers; important in biomedical applications. Since the initial report by Hedrick and co-workers that 4-dimethylaminopyridine (DMAP) was an effective catalyst for the ROP of LA,20 several classes of organic catalyst have been reported including other nucleophilic catalysts such as phosphines21 or N-heterocyclic carbenes,22–25 supramolecular catalysts such as thiourea,26,27 fluorinated alcohol,28 (thio)amidoindoles29,30 or sulfonamides31 in combination with tertiary amines, basic catalysts including amidines,32guanidines,32–34 aminothiazolines35 and phosphazenes36,37 or acid catalysts including trifluoromethanesulfonic acid and methanesulfonic acid.38,39 Amongst these catalysts thiourea/tertiary amine catalysts, 1 (Fig. 1) have been shown to exert excellent levels of control over ROP of lactide with minimal transesterification side-reactions, resulting from selective activation of cyclic rather than linear esters.26,27
 | ||
| Fig. 1 Chemical structures of thiourea/tertiary amine catalyst for the ring-opening polymerization of lactide, 1, and astaxanthin, 2. | ||
Astaxanthin, 2 (Fig. 1), is a highly conjugated carotenoid that in common with other compounds of this genre is only synthesised in nature by plants, phytoplankton, algae and some species of bacteria and fungi.40,41 Its primary biological function is to either act as an antioxidant or aid light absorption during photosynthesis.40,41 As a consequence of these properties, astaxanthin and other carotenoids have been shown to possess many potentially beneficial uses in anti-cancer therapies,42,43neuroprotection,44 and against skin photosensitivity.45 Amongst the leading examples are Cardax; a disodium disuccinate functionalised version of astaxanthin used as an anti-inflammatory for the treatment of cardiovascular disease,46,47astaxanthin tetrasodium diphosphate for use in cancer chemoprevention,48 and astaxanthin dilysine tetrahydrochloride for improved targeting in cancer and cardiovascular disease treatment.49–51Astaxanthin is also one of the main carotenoids responsible for the pink colouration observed in the flesh pigmentation of salmonids and shellfish, a highly prised asset for consumers52–54 that is also essential for the general health and wellbeing of the fish.41 However, the inability of these species to synthesise carotenoids leads to a reliance on dietary uptake in order to exploit them as pigmentation sources.41,55 The scarce availability of such sources to farmed salmon requires that astaxanthin is fed in a synthetic form, however, such colour feeding is expensive and inefficient. Astaxanthin disuccinate esters have been shown to increase both the stability of the native astaxanthin and aids digestive retention.56
Utilization of residual alcohol groups on bioactive molecules, such as those present in native astaxanthin, to initiate ROP may thus lead to a simple strategy for controlled release encapsulation. With the utilization of highly selective organic catalysis, it is expected that PLAs can be prepared with very high levels of control and end-group fidelity thus retaining high and predictable levels of bioactive molecule incorporation. Astaxanthin is applied as an exemplar for a far wider set of compounds.
Experimental
Materials
rac-Lactide (Aldrich) was purified by recrystallization from dry dichloromethane and sublimation two times before use. Dichloromethane and CDCl3 were refluxed over CaH then distilled, degassed and stored under a nitrogen atmosphere. Astaxanthin and 4-pyrene-1-butanol were obtained from Aldrich, both initiators were dried over activated molecular sieves in a dichloromethane solution. All other chemicals and solvents were obtained from Aldrich and used as received.General considerations
All manipulations were performed under moisture and oxygen free conditions either in a nitrogen filled glovebox or by standard Schlenk techniques. Mass spectra were acquired by MALDI-TOF (matrix-assisted laser desorption and ionization time-of-flight) mass spectrometry using a Bruker Daltonics Ultraflex II MALDI-TOF mass spectrometer, equipped with a nitrogen laser delivering 2 ns laser pulses at 337 nm with positive ion TOF detection performed using an accelerating voltage of 25 kV. DCTB (trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene]malononitrile) was used as a matrix (50 mg mL−1) with NaI as cationization agent (150 mg mL−1). All samples were taken directly from previous NMR samples in CDCl3 and spotted sequentially on a ground steel plate. Spectra were recorded using Bruker Flex Control software. Gel-permeation chromatography (GPC) was used to determine the molecular weights and PDIs of the synthesized polymers and was performed in either CHCl3 or THF. CHCl3 analysis was performed on a Polymer Laboratories modular GPC system comprising of a Polymer Laboratories Midas autosampler and LC1120 HPLC pump equipped with a guard column (Polymer Laboratories PLGel 5 µM, 50 × 7.5 mm), two mixed D columns (Polymer Laboratories PLGel 5 µM, 300 × 7.5 mm) and a Polymer Laboratories ERC-7515A differential refractive index (DRI) detector. The mobile phase was chloroform/triethyl amine (95/5) eluent at a flow rate of 1.0 mL min−1 and samples were calibrated against linear poly(methyl methacrylate) standards obtained from Polymer Laboratories using Cirrus v3.0; elution time was standardized against that of toluene. UV/Vis detected GPC analysis was performed on a Polymer Laboratories PL-GPC50 unit equipped with a guard column (Polymer Laboratories PLGel 5 µM, 50 × 7.5 mm), two mixed D columns (Polymer Laboratories PLGel 5 µM, 300 × 7.5 mm), a Polymer Laboratories ERC-7515A differential refractive index (DRI) detector and a Shimadzu SPD-6AV UV detector set to record at 480 nm. The mobile phase was THF/triethyl amine (95/5) eluent at a flow rate of 1.0 mL min−1 and samples were calibrated against linear poly(methyl methacrylate) standards obtained from Polymer Laboratories using Cirrus v3.0; elution time was standardized against that of toluene. IR spectra were collected from solids on a Bruker Vector 22 FTIR spectrometer with a golden gate cell and analyzed using Opus. 1H and 13C NMR spectra were recorded on a Bruker DPX-400 spectrometer at 293 K unless stated. Chemical shifts are reported as δ in parts per million (ppm) and referenced to the chemical shift of the residual solvent resonances (CDCl31H: δ = 7.26 ppm; 13C: δ = 77.16 ppm).General procedure for astaxanthin–poly(lactide) synthesis
All lactide polymerisations were carried out in a nitrogen filled glovebox. Astaxanthin, thiourea/amine catalyst, 2 (1:0.1 w.r.t. OH group), and rac-lactide were weighed into a vial and dichloromethane added (10% solids). The reaction was stirred at ambient temperature until >95% conversion (1H NMR) was reached. The reaction was removed from the glovebox, and the polymer filtered through a short silica frit, using dichloromethane:methanol (95:5) as eluent to remove the thiourea/amine catalyst. The solution was concentrated in vacuo and precipitated into petroleum ether (bp 60–80 °C). The distinctive red coloured polymer was collected by vacuum filtration and dried in a vacuum oven (40 °C) overnight.
General procedure for astaxanthin and 4-pyrene-1-butanol initiated poly(lactide) transesterification study
The polymerisations were carried out in a nitrogen filled glovebox. Alcohol, thiourea/amine catalyst, 2 (1:0.1 per alcohol), and rac-lactide (0.5 g, 0.03 mol) were weighed into a vial and dichloromethane or CDCl3 added (5 mL). The reactions were stirred at ambient temperature and sampled regularly for MALDI-TOF analysis of the transesterification products.
Results and discussion
A series of poly(lactide)s were prepared by the ROP of L-LA catalyzed by the thiourea/tertiary amine catalyst, 1, using astaxanthin as a bifunctional initiating species. Good control of the polymerization was achieved (Table 1) such that PLAs with molecular weights close to those targeted from the monomer-to-initiator ratio ranging from 2500 to 30000 g mol−1 (as determined by GPC analysis) were obtained. While at lower degrees of polymerization (DPs) the polymers display slightly higher polydispersities, samples of higher molecular weight are noted to narrow, consistent with many controlled polymerizations as a consequence of the percentage difference between chain sizes becoming nominal.1,43 Monitoring of the monomer conversion and Mn of the polymer with time in these polymerizations revealed that the polymerizations proceed with first order kinetics (see ESI†) while displaying a linear correlation between Mn and % monomer conversion (Fig. 2), which indicates that the polymerization is well controlled. It is noteworthy that slight deviations from first order kinetics occur at increased time periods that is tentatively attributed to transesterification side reactions (see below).
![Chart of Mn and PDI versus % monomer conversion for the astaxanthin-initiated polymerization of lactide catalyzed by 1 (R = [monomer]/[initiator]).](/image/article/2011/PY/c0py00227e/c0py00227e-f2.gif) | ||
| Fig. 2 Chart of Mn and PDI versus % monomer conversion for the astaxanthin-initiated polymerization of lactide catalyzed by 1 (R = [monomer]/[initiator]). | ||
Demonstration that the polymerization was initiated from astaxanthin was obtained by GPC, NMR and IR spectroscopic analysis of the resultant polymers. As a consequence of high levels of conjugation in astaxanthin, a strong absorption in its UV/Vis spectrum can be observed at λ = 480 nm. Analysis of the astaxanthin-initiated polymers by UV/Vis-detected GPC shows a strong absorbance at a shifted retention time compared to free astaxanthin (Fig. 3). These data demonstrate that the astaxanthin is distributed throughout the polymer. IR spectroscopic analysis revealed no evidence of the original astaxanthin OH peak at 3400 cm−1 and while the very strong signal for the carbonyl groups of the poly(lactide) at 1750 cm−1 masks the shifted signal of the astaxanthin carbonyls, the peak that corresponds to the hydrocarbon backbone of astaxanthin at approximately 3000 cm−1 is still observed in the polymer spectrum, in agreement with the presence of the astaxanthin in the poly(lactide). Further analysis of the polymers by 1H NMR spectroscopy (Fig. 4a) revealed a shift of resonances corresponding to the astaxanthin protons upon polymer chain growth, most notably in the resonances attributed to those adjacent to the initiating alcohol (3) from δ = 4.35 ppm to δ = 5.53 ppm upon esterification. Finally, the MALDI-TOF spectrum of the DP50 astaxanthin-initiated PLA (Fig. 4b) shows a mass distribution centred at 8993.0 Da which corresponds to a sodium-charged, astaxanthin-initiated PLA with a total DP = 58 (calculated mass = 8977.6 Da). Notably, the major repeat unit is 144 Da. The absence of a significant distribution spaced by 72 Da indicates that the level of transesterification is low.
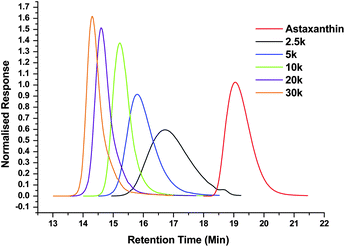 | ||
| Fig. 3 UV/Vis-detected GPC traces for astaxanthin and astaxanthin-initiated poly(lactide)s. | ||
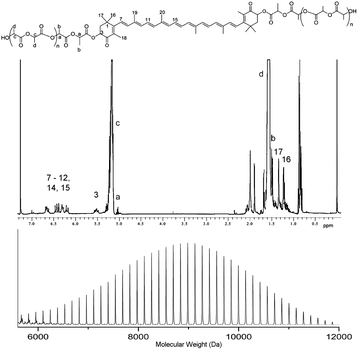 | ||
| Fig. 4 (a) 1H NMR spectrum (CDCl3, 400 MHz) and (b) MALDI-TOF spectrum of DP35 astaxanthin-initiated poly(lactide). | ||
During the course of our studies we noted that samples that were left for extended time periods prior to isolation led to the observation of increased distributions spaced by 72 Da after MALDI-TOF analysis. Such observations contrast previous studies of lactide polymerization using this catalyst system in which transesterification products are negligible.26,27 Indeed upon exposure of methyl benzoate and either ethanol or isopropanol in the presence of 5 mol% 1, the quantitative recovery of the methyl benzoate was exclusively observed in both cases after 48 hours.26 Our data indicate that transesterification does occur and as such we directed further studies to further investigate these observations.
A series of lactide polymerizations (target DP = 50 total) were performed, catalyzed by 1 with initiation from astaxanthin or 4-pyrene-1-butanol as a control; samples were taken periodically over a 36 day period. In both cases, polymerizations were well controlled, however, GPC analysis of the polymers revealed that the astaxanthin-initiated polymers displayed slightly broader PDIs, however, even at extended time periods, PDIs did not broaden significantly (see ESI†). Analysis of these polymers by MALDI-TOF, however, revealed that significant transesterification occurs (Fig. 5). Comparison of MALDI-TOF spectra taken at various time intervals clearly shows that after 1 day (>95% monomer conversion) the dominant distribution is attributable to a 144 Da spacing which indicates that only minimal transesterification is observed. At increasing time periods the distribution is observed to broaden with a second distribution spaced by 72 Da increasing in intensity such that after 21 days noticeable broadening of the distribution is observed with peaks spaced by 72 Da roughly at half intensity to the main 144 Da spaced distribution.
 | ||
| Fig. 5 Comparison of MALDI-TOF mass spectra between 6 hours and 36 days in the ring-opening polymerization of lactide (target DP = 50; 25 per arm) initiated from astaxanthin catalyzed by 1. | ||
Noticeably, however, initiation of lactide ROP from 4-pyrene-1-butanol results in greatly increased levels of control over the polymerization with respect to reduced transesterification side reactions at increased time periods. MALDI-TOF MS analysis of this polymerization (Fig. 6) reveals that after 21 days there are no apparent signs of transesterification product in the MALDI spectrum, as evidenced by a lack of significant distribution spaced by 72 Da; even after 36 days the amount of transesterified polymer is very low, notably much reduced compared to that observed for the astaxanthin-initiated polymer.
 | ||
| Fig. 6 Comparison of MALDI-TOF mass spectra after 21 days and 36 days in the ring-opening polymerization of lactide (target DP = 50) initiated from 4-pyrene-1-butanol catalyzed by 1. | ||
While both polymers have an overall DP = 50, the bifunctional astaxanthin-initiated sample has two alcohol chain-ends compared to the 4-pyrene-1-butanol-initiated sample that only contains one. In order to confirm that this difference was not simply a consequence of the alcohol chain-end-to-polymer ratio, a DP25 PLA (ca. 3500 g mol−1) initiated from 4-pyrene-1-butanol was prepared and studied. At extended time periods, this polymer displayed a good agreement with the observations for the longer polymer such that while MALDI-TOF analysis reveals a moderate broadening of the molecular weight distribution and a negligible amount of transesterified product is observed after 36 days (see ESI†). These data suggest that the presence of two alcohol end-groups on a single polymer chain leads to decreased resistance to transesterification side-reactions. To confirm this observation, ROP of lactide initiated by a simple diol, namely 1,3-propane diol, under identical conditions to those described for initiation from astaxanthin was investigated. In common with the astaxanthin-initiated ROP, monitoring the polymerization reaction over 27 days led to the observation of significant transesterification of the polymers, evidenced by a notable distribution spaced by 72 Da. These data are consistent with the increased transesterification being a consequence of the bifunctional nature of the initiation (and hence propagating) species. Notably, at increased time periods GPC analysis of the polymers revealed not only a broadening of the PDI (from 1.12 after 1 day to 1.23 after 27 days) but the presence of low molecular weight species suspected to be cyclic oliomers.
Conclusions
Astaxanthin-containing poly(lactide)s were successfully obtained by the organocatalytic ROP of lactide from both alcohol groups on astaxanthin using a thiourea/tertiary amine bifunctional catalyst, thus demonstrating the utility of this simple approach for the preparation of a wide range of polymer conjugates. Notably, comparison of the polymerization reactions at extended time periods up to 36 days against those initiated from the monofunctional alcohol 4-pyrene-1-butanol, revealed that the bifunctional initiator led to greatly increased levels of transesterification, likely a consequence of increased intramolecular transesterification side reactions.Acknowledgements
The Research Councils UK (RCUK) are acknowledged for funding a fellowship to A.P.D. We gratefully acknowledge the support provided by EPSRC (EP/C007999/1) for the purchase of the Bruker Ultraflex MALDI-TOF MS instrument. We thank Warwick Effect Polymers Ltd and EPSRC for funding (H.M.).Notes and references
- M. C. Branco and J. P. Schneider, Acta Biomater., 2009, 5, 817–831 CrossRef CAS.
- D. Roy, J. N. Cambre and B. S. Sumerlin, Prog. Polym. Sci., 2010, 35, 278–301 CrossRef CAS.
- S. Cartmell, J. Pharm. Sci., 2009, 98, 430–441 CrossRef CAS.
- C. Wischke and S. P. Schwendeman, Int. J. Pharm., 2008, 364, 298–327 CrossRef CAS.
- U. Edlund and A. C. Albertsson, Adv. Polym. Sci., 2002, 157, 67–112 CAS.
- A. P. Gupta and V. Kumar, Eur. Polym. J., 2007, 43, 4053–4074 CrossRef CAS.
- X. L. Hu and X. B. Jing, Expert Opin. Drug Delivery, 2009, 6, 1079–1090 Search PubMed.
- E. Rytting, J. Nguyen, X. Y. Wang and T. Kissel, Expert Opin. Drug Delivery, 2008, 5, 629–639 Search PubMed.
- K. E. Uhrich, S. M. Cannizzaro, R. S. Langer and K. M. Shakesheff, Chem. Rev., 1999, 99, 3181–3198 CrossRef CAS.
- J. T. F. Keurentjes, M. F. Kemmere, H. Bruinewoud, M. Vertommen, S. A. Rovers, R. Hoogenboom, L. F. S. Stemkens, F. Peters, N. J. C. Tielen, D. T. A. van Asseldonk, A. F. Gabriel, E. A. Joosten and M. A. E. Marcus, Angew. Chem., Int. Ed., 2009, 48, 9867–9870 CrossRef CAS.
- B. Le Droumaguet and J. Nicolas, Polym. Chem., 2010, 1, 563–598 RSC.
- O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef.
- A. P. Dove, Chem. Commun., 2008, 6446–6470 RSC.
- N. E. Kamber, W. Jeong, R. M. Waymouth, R. C. Pratt, B. G. G. Lohmeijer and J. L. Hedrick, Chem. Rev., 2007, 107, 5813–5840 CrossRef CAS.
- R. H. Platel, L. M. Hodgson and C. K. Williams, Polym. Rev., 2008, 48, 11–63 Search PubMed.
- M. J. Stanford and A. P. Dove, Chem. Soc. Rev., 2010, 39, 486–494 RSC.
- A. C. Albertsson and R. K. Srivastava, Adv. Drug Delivery Rev., 2008, 60, 1077–1093 CrossRef CAS.
- J. Kadokawa and S. Kobayashi, Curr. Opin. Chem. Biol., 2010, 14, 145–153 CrossRef CAS.
- M. K. Kiesewetter, E. J. Shin, J. L. Hedrick and R. M. Waymouth, Macromolecules, 2010, 43, 2093–2107 CrossRef CAS.
- F. Nederberg, E. F. Connor, M. Moller, T. Glauser and J. L. Hedrick, Angew. Chem., Int. Ed., 2001, 40, 2712–2715 CrossRef CAS.
- M. Myers, E. F. Connor, T. Glauser, A. Mock, G. Nyce and J. L. Hedrick, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 844–851 CrossRef CAS.
- E. F. Connor, G. W. Nyce, M. Myers, A. Mock and J. L. Hedrick, J. Am. Chem. Soc., 2002, 124, 914–915 CrossRef CAS.
- O. Coulembier, A. P. Dove, R. C. Pratt, A. C. Sentman, D. A. Culkin, L. Mespouille, P. Dubois, R. M. Waymouth and J. L. Hedrick, Angew. Chem., Int. Ed., 2005, 44, 4964–4968 CrossRef CAS.
- S. Csihony, D. A. Culkin, A. C. Sentman, A. P. Dove, R. M. Waymouth and J. L. Hedrick, J. Am. Chem. Soc., 2005, 127, 9079–9084 CrossRef CAS.
- A. P. Dove, R. C. Pratt, B. G. G. Lohmeijer, D. A. Culkin, E. C. Hagberg, G. W. Nyce, R. M. Waymouth and J. L. Hedrick, Polymer, 2006, 47, 4018–4025 CrossRef CAS.
- A. P. Dove, R. C. Pratt, B. G. G. Lohmeijer, R. M. Waymouth and J. L. Hedrick, J. Am. Chem. Soc., 2005, 127, 13798–13799 CrossRef CAS.
- R. C. Pratt, B. G. G. Lohmeijer, D. A. Long, P. N. P. Lundberg, A. P. Dove, H. B. Li, C. G. Wade, R. M. Waymouth and J. L. Hedrick, Macromolecules, 2006, 39, 7863–7871 CrossRef CAS.
- O. Coulembier, D. R. Sanders, A. Nelson, A. N. Hollenbeck, H. W. Horn, J. E. Rice, M. Fujiwara, P. Dubois and J. L. Hedrick, Angew. Chem., Int. Ed., 2009, 48, 5170–5173 CrossRef CAS.
- S. Koeller, J. Kadota, A. Deffieux, F. Peruch, S. Massip, J. M. Leger, J. P. Desvergne and B. Bibal, J. Am. Chem. Soc., 2009, 131, 15088–15089 CrossRef CAS.
- S. Koeller, J. Kadota, F. Peruch, A. Deffieux, N. Pinaud, I. Pianet, S. Massip, J. M. Leger, J. P. Desvergne and B. Bibal, Chem.–Eur. J., 2010, 16, 4196–4205 CrossRef CAS.
- A. Alba, A. Schopp, A. P. D. Delgado, R. Cherif-Cheikh, B. Martin-Vaca and D. Bourissou, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 959–965 CrossRef CAS.
- B. G. G. Lohmeijer, R. C. Pratt, F. Leibfarth, J. W. Logan, D. A. Long, A. P. Dove, F. Nederberg, J. Choi, C. Wade, R. M. Waymouth and J. L. Hedrick, Macromolecules, 2006, 39, 8574–8583 CrossRef CAS.
- R. C. Pratt, B. G. G. Lohmeijer, D. A. Long, R. M. Waymouth and J. L. Hedrick, J. Am. Chem. Soc., 2006, 128, 4556–4557 CrossRef CAS.
- L. Zhang, R. C. Pratt, F. Nederberg, H. W. Horn, J. E. Rice, R. M. Waymouth, C. G. Wade and J. L. Hedrick, Macromolecules, 2010, 43, 1660–1664 CrossRef CAS.
- J. M. Becker, S. Tempelaar, M. J. Stanford, R. J. Pounder, J. A. Covington and A. P. Dove, Chem.–Eur. J., 2010, 16, 6099–6105 CAS.
- L. Zhang, F. Nederberg, J. M. Messman, R. C. Pratt, J. L. Hedrick and C. G. Wade, J. Am. Chem. Soc., 2007, 129, 12610–12611 CrossRef CAS.
- L. Zhang, F. Nederberg, R. C. Pratt, R. M. Waymouth, J. L. Hedrick and C. G. Wade, Macromolecules, 2007, 40, 4154–4158 CrossRef CAS.
- D. Bourissou, B. Martin-Vaca, A. Dumitrescu, M. Graullier and F. Lacombe, Macromolecules, 2005, 38, 9993–9998 CrossRef CAS.
- H. R. Kricheldorf and R. Dunsing, Makromol. Chem., 1986, 187, 1611–1625 CrossRef CAS.
- B. Demmig-Adams, A. M. Gilmore and W. W. Adams, FASEB J., 1996, 10, 403–412 CAS.
- J. Higuera-Ciapara, L. Felix-Valenzuela and F. M. Goycoolea, Crit. Rev. Food Sci. Nutr., 2006, 46, 1549–7852.
- P. Palozza, Curr. Pharmacogenomics, 2004, 2, 35–45 Search PubMed.
- T. A. D. Smith, Br. J. Biomed. Sci., 1998, 55, 268–275 Search PubMed.
- F. J. Romero, F. Bosch-Morrell and M. Miranda, US Pat., 2006205826, 2006.
- M. M. Mathews-Roth, Int. J. Dermatol., 1979, 18, 835–836 CrossRef CAS.
- B. J. Foss, H. R. Sliwka, V. Partali, S. N. Naess, A. Elgsaeter, T. B. Melo, K. R. Naqvi, S. O'Malley and S. F. Lockwood, Chem. Phys. Lipids, 2005, 135, 157–167 CrossRef CAS.
- D. A. Frey, E. W. Kataisto, J. L. Ekmanis, S. O'Malley and S. F. Lockwood, Org. Process Res. Dev., 2004, 8, 796–801 Search PubMed.
- L. M. Hix, D. A. Frey, M. D. McLaws, M. Osterlie, S. F. Lockwood and J. S. Bertram, Carcinogenesis, 2005, 26, 1634–1641 CrossRef CAS.
- H. L. Jackson, A. J. Cardounel, J. L. Zweier and S. F. Lockwood, Bioorg. Med. Chem. Lett., 2004, 14, 3985–3991 CrossRef CAS.
- S. N. Naess, H. R. Sliwka, V. Partali, T. B. Melo, K. R. Naqvi, H. L. Jackson and S. F. Lockwood, Chem. Phys. Lipids, 2007, 148, 63–69 CrossRef CAS.
- F. Zsila, I. Fitos, Z. Bikadi, M. Simonyi, H. L. Jackson and S. F. Lockwood, Bioorg. Med. Chem. Lett., 2004, 14, 5357–5366 CrossRef CAS.
- T. R. Gormley, Ir. J. Agric. Food Res., 1992, 31, 199–202 Search PubMed.
- R. C. Rounds, C. L. Glenn and A. O. Bush, J. Food Sci., 1992, 57, 572–574 CrossRef.
- D. I. Skonberg, R. W. Hardy, F. T. Barrows and F. M. Dong, Aquaculture, 1998, 166, 269–277 CrossRef.
- T. W. Goodwin, Annu. Rev. Nutr., 1986, 6, 273–297 CrossRef CAS.
- A. Gloor and W. Simon, US Pat., 7253297, 2007.
Footnote |
| † Electronic supplementary information (ESI) available: Additional data, charts and MALDI-TOF spectra. See DOI: 10.1039/c0py00227e |
| This journal is © The Royal Society of Chemistry 2011 |