Thiol–yne ‘click’ chemistry as a route to functional lipid mimetics†‡
Sandeep S.
Naik
,
Justin W.
Chan
,
Christopher
Comer
,
Charles E.
Hoyle
and
Daniel A.
Savin
*
The University of Southern Mississippi, School of Polymers and High Performance Materials, 118 College Drive #5050, Hattiesburg, MS 39406, USA. E-mail: daniel.savin@usm.edu; Fax: +1-601-266-5504; Tel: +1-601-266-5395
First published on 7th September 2010
Abstract
Thiol–alkyne ‘click’ chemistry is a modular, efficient mechanism to synthesize complex A2B 3-arm star polymers. This general motif is similar to a phospholipid where the A blocks correspond to lypophilic chains and the B block represents the polar head group. In this communication we employ thiol–yne chemistry to produce polypeptide-based A2B lipid mimetics. The utility of the thiol–yne reaction is demonstrated by using a divergent and a convergent approach in the synthesis. These polymers self-assemble in aqueous solution into spherical vesicles with a relatively narrow size distribution independent of block composition over the range studied. Using the thiol–yne convergent synthesis, we envision a modular approach to functionalize proteins or oligopeptides with lipophilic chains that can imbed seamlessly into a cell membrane.
Lipid mimetics are synthetic molecules that resemble naturally occurring phospholipids, the basic building blocks of the cell membrane. Cell membranes contain various membrane proteins which are responsible for signaling and transport. In order to get a thorough understanding of these membrane proteins, they must be studied in a native environment. Synthetic lipid bilayers (liposomes), based on lipid like small-molecule amphiphiles, have been extensively studied.1–4 Liposomes, however, lack toughness and stability for a wide range of reaction parameters and conditions. This problem is overcome through the use of polymersomes, which are vesicles composed of amphiphilic block copolymers.5–9 Typically, these polymersomes are diblock copolymers whereby the equilibrium morphology is dictated roughly by the relative volumes of the hydrophobic and hydrophilic blocks. The utility of the proposed A2B motif ensures that the hydrophobic volume is maximized, favoring bilayer formation. Polypeptide-based polymersomes are a useful class of materials whereby well-defined secondary structure changes lead to pH and/or temperature responsive, biocompatible structures.10–14
The most common method to prepare polypeptide-based block copolymers invokes the ring-opening polymerization of the peptide N-carboxyanhydride (NCA) using a polymer macroinitiator.15 An alternative method is to couple preformed polypeptides with polymer blocks using various coupling strategies. The use of ‘click’ chemistry, in particular the copper-catalyzed alkyne–azide cycloaddition, has been proven to be very beneficial in this regard by giving opportunities to synthesize various molecular geometries and compositions which are cumbersome using conventional techniques.16–20
Radical-mediated thiol–alkene chemistry is another such ‘click’ reaction;21–25 however, it has only been recently explored as a coupling technique for polypeptides.26–29 Of the same family is thiol–alkyne chemistry, which results in 3-arm star materials via sequential thiol–ene additions at the same reaction center.30–33 Similar to the thiol–ene reaction, the thiol–yne reaction proceeds rapidly and efficiently under ambient conditions without the use of a catalyst (i.e.copper) that may limit its usefulness in biomedical applications. The use of thiol–yne chemistry for peptide conjugation is very recent.34,35 In this communication, we exploit this 3-arm star motif to produce complex, lipid-mimetic polymersomes.36–38
Herein we report the synthesis and solution characterization of multiply responsive, polypeptide-based 3-arm star materials that serve as lipid mimetic molecules. These materials contain a peptide block, poly(lysine) (PK) or poly(glutamic acid) (PE), as the head group along with two long alkyl chain ‘legs’. Two different routes were used to synthesize these lipid mimetics (Fig. 1). In the divergent synthesis, thiol–yne chemistry is used to functionalize propargyl amine (PA), which is then used to initiate the NCA ring-opening polymerization of benzyl glutamate or Z-lysine NCA. In the convergent synthesis, PA-initiated poly(benzyl glutamate) is functionalized with dodecanethiol (DDT) legs viathiol–yne chemistry. Prior to deprotection, DDT2–P(Z-Lys) is able to self-assemble into gels in organic (i.e.tetrahydrofuran and chloroform) media. Upon deprotection, all samples self-assemble in aqueous media into bilayer vesicles. Using the thiol–yne convergent synthesis, we envision a modular approach to functionalize proteins or oligopeptides with lipophilic chains to imbed seamlessly into a cell membrane.
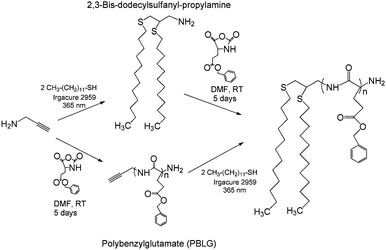 | ||
| Fig. 1 Divergent (top) and convergent (bottom) routes to DDT2–PBLG 3-arm star polymers. Although this is shown for PBLG polymers, the same protocol can be used for DDT2–P(Z-Lys). | ||
Divergent synthesis of DDT2–PK22 and DDT2–PE16
In the divergent approach, 2–4 eq. of DDT was reacted with 1 eq. of PA viathiol–yne chemistry in the presence of photo-initiator under atmospheric conditions to afford 2,3-bis-dodecylsulfanyl-propylamine (DDT2–NH2). This is a two step reaction process: (1) during the first step, one thiol adds to the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bond forming an intermediate vinyl thioether. (2) This further reacts with second thiol to yield 1,2-bis-addition product. Bowman had shown previously that the second addition occurs with a rate ca. 3 times that of the first addition for aliphatic alkynes.31 The reaction was monitored by real time FTIR, tracking the disappearance of peaks corresponding to thiol (2570 cm−1) and alkyne (2120 cm−1 and 3300 cm−1) moieties (ESI‡). Reacting the PA in neat thiol yielded 50% conversion after approximately 10 minutes. The addition of benzene solvent (ca. 50% v/v) caused the conversion to improve to 80%, leading us to conclude that lack of mobility hinders complete reaction in the neat mixture. When the thiol
C bond forming an intermediate vinyl thioether. (2) This further reacts with second thiol to yield 1,2-bis-addition product. Bowman had shown previously that the second addition occurs with a rate ca. 3 times that of the first addition for aliphatic alkynes.31 The reaction was monitored by real time FTIR, tracking the disappearance of peaks corresponding to thiol (2570 cm−1) and alkyne (2120 cm−1 and 3300 cm−1) moieties (ESI‡). Reacting the PA in neat thiol yielded 50% conversion after approximately 10 minutes. The addition of benzene solvent (ca. 50% v/v) caused the conversion to improve to 80%, leading us to conclude that lack of mobility hinders complete reaction in the neat mixture. When the thiol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :alkyne ratio is increased to 4:1 (in benzene), the reaction becomes quantitative, achieving 95% conversion approximately three minutes. Our original hypothesis was that there is competition between the radical addition of the thiol to the acetylene group and the acid–base reaction between the amine of PA and thiol of DDT. To test this, we performed an experiment similar to Fairbanks et al. and compared the FTIR spectrum of DDT (1.9 M in benzene) in the presence and absence of PA (2.5 M) and found that the intensity of the thiol stretch remained unchanged.39 Nonetheless, from the FTIR conversion data and 1H NMR spectra (Fig. 2, middle), it is clear that under these conditions the bis-addition was successful due to the presence of aliphatic chains in the 1–2 ppm region. In addition, the 13C NMR (ESI‡) only shows the bis-addition product.
:alkyne ratio is increased to 4:1 (in benzene), the reaction becomes quantitative, achieving 95% conversion approximately three minutes. Our original hypothesis was that there is competition between the radical addition of the thiol to the acetylene group and the acid–base reaction between the amine of PA and thiol of DDT. To test this, we performed an experiment similar to Fairbanks et al. and compared the FTIR spectrum of DDT (1.9 M in benzene) in the presence and absence of PA (2.5 M) and found that the intensity of the thiol stretch remained unchanged.39 Nonetheless, from the FTIR conversion data and 1H NMR spectra (Fig. 2, middle), it is clear that under these conditions the bis-addition was successful due to the presence of aliphatic chains in the 1–2 ppm region. In addition, the 13C NMR (ESI‡) only shows the bis-addition product.
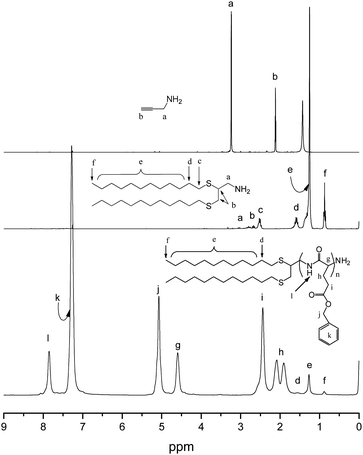 | ||
| Fig. 2 1H NMR for the divergent synthesis of DDT2–PBLG lipid mimetics: top, PA; middle, DDT2–NH2; bottom, DDT2–PBLG. | ||
Z-Lysine NCA and benzyl glutamate NCA (BLG NCA) were synthesized using the Fuch–Farthings method. DDT2–NH2 was then used to initiate the ring-opening polymerization of the NCAs (target DPn = 20 for both polymers). For the synthesis of DDT2–PBLG, the 1H NMR (Fig. 2, bottom) confirms the presence of the phenyl protecting groups in the region around 7–7.5 ppm, and a suppression of the aliphatic chains in intensity relative to the PBLG peaks.
When the lysine side-chains in DDT2–P(Z-Lys)22 are protected, this star polymer self-assembles into organogels in CHCl3 and THF (Fig. 3). Gels in CHCl3 (10 wt%) are qualitatively strong and resistant to moderate sonication compared with gels in THF. Consistent with previous studies of P(Z-Lys) containing polymers, organogelation is likely due to intermolecular β-sheet formation between chains.40 The polypeptide blocks were then deprotected to render the resulting A2B star polymer water soluble. A lipopolypeptide similar to DDT2–P(Z-Lys) was synthesized previously by Dimitrov using a 3-step protocol utilizing the thiol-Michael addition reaction for the preparation of the DDT2–NH2 macroinitiator.38 The thiol–yne approach presented here is more facile and modular in the choice of lipophilic groups.
 | ||
| Fig. 3 Organogel from DDT2–P(Z-Lys)22 in 10 wt% CHCl3. | ||
Convergent synthesis of DDT2–PE10
PA was used to initiate the ring-opening polymerization of BLG NCA (target DPn = 10) to yield acetylene-terminated PBLG. The purified product was then reacted with 4 eq. of dodecanethiolviathiol–yne chemistry. The polymer was further purified and deprotected as above to yield DDT2–PE10 star polymer. 1H NMR was used to confirm the completion of reaction and characterize the intermediate products, and end group analysis was used to determine the degree of polymerization of the peptide blocks (ESI‡). From gel permeation chromatography in DMF, it was determined that the polydispersities of the A2B star polymers were between 1.3 and 1.5, typical for NCA ring-opening polymerizations under standard conditions.Upon deprotection, all of the synthesized polymers self-assemble in aqueous solution. These A2B star polymers were characterized using dynamic (DLS) and static light scattering (SLS) techniques, as well as transmission electron microscopy (TEM). Fig. 4 shows a plot of the average decay rate of the DLS autocorrelation function (from the second cumulant) as a function of the square of the scattering vector q for the three A2B star polymers synthesized. The linearity suggests that the scattering comes primarily from the Brownian motion of spherical particles. The slope is then equal to the diffusion coefficient, and the hydrodynamic radius (Rh) of the aggregates is calculated using the Stokes–Einstein relation. For all polymers, the normalized variance (polydispersity) in decay rate was ca. 0.08–0.13, suggesting a relatively narrow size distribution of particles. The radius of gyration (Rg) was determined from SLS using Zimm analysis (ESI‡), and the ratio ρ = Rg/Rh is used as an indicator of morphology.
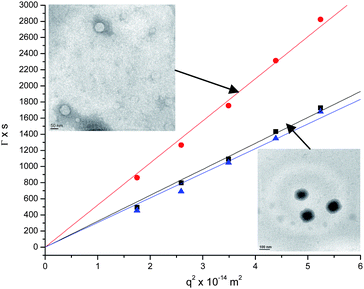 | ||
| Fig. 4 Average decay rate of the DLS autocorrelation function vs.q2 for determination of Dm and Rh from DLS data. Linearity suggests Brownian diffusion of spherical particles. Insets: TEM images of DDT2–PK22 (top) and DDT2–PE16 (bottom). Scale bars are 50 and 100 nm respectively. | ||
For DDT2–PE16 and DDT2–PK22 synthesized via the divergent approach, the Rh was 76 (black squares) and 47 nm (red circles) respectively. The corresponding Rgs were 83 and 49 nm, leading to ρ values of 1.09 and 1.04, respectively, close to the theoretical value of 1.0 for a vesicle morphology. For DDT2–PE10 (blue triangles) synthesized via the convergent approach, Rh and Rg were 80 and 75 nm, respectively, leading to ρ = 0.94, again close to unity. The inset of Fig. 4 shows representative TEM images for DDT2–PK22 and DDT2–PE16. It is clear that the lysine-containing polymer exhibits vesicle morphology with a size consistent with the light scattering data. The morphology is less pronounced in DDT2–PE in part due to the acidic stain. This results in a higher contrast in the core of the vesicle and a lighter membrane, as shown above.
In summary, we have demonstrated the use of thiol–alkyne ‘click’ chemistry as an efficient and rapid tool to synthesize multiply responsive Y-shaped architectures which serve as lipid mimetics. Due to the highly modular, tailorable, simplistic approach in the synthesis, these bilayer structures will have many applications and help toward the systematic study of membranes in tunable environments. This chemistry holds potential for designing more interesting molecular architectures currently inaccessible through time consuming and tedious conventional polymerization techniques.
Acknowledgements
This communication is dedicated to the late Prof. Charlie Hoyle. The authors would like to thank Prof. Charles McCormick for assistance with GPC. Funding was provided by USM startup funds and by the NSF DMR MRI-0421406 for the purchase of the JEOL JEM-2100 electron microscope.Notes and references
- M. G. Elferink, J. G. de Wit, R. Demel, A. J. Driessen and W. N. Konings, J. Biol. Chem., 1992, 267, 1375–1381 CAS.
- M. T. Paternostre, M. Roux and J. L. Rigaud, Biochemistry, 1988, 27, 2668–2677 CrossRef CAS.
- A. M. Seddon, P. Curnow and P. J. Booth, Biochim. Biophys. Acta, Biomembr., 2004, 1666, 105–117 CrossRef CAS.
- J. Watanabe, Y. Asaka, K. Mino and S. Kanamura, J. Electron Microsc., 1996, 45, 171–176 CAS.
- B. M. Discher, D. A. Hammer, F. S. Bates and D. E. Discher, Curr. Opin. Colloid Interface Sci., 2000, 5, 125–131 CrossRef CAS.
- B. M. Discher, Y.-Y. Won, D. S. Ege, J. C. M. Lee, F. S. Bates, D. E. Discher and D. A. Hammer, Science, 1999, 284, 1143–1146 CrossRef.
- J. C. M. Lee, H. Bermudez, B. M. Discher, M. A. Sheehan, Y.-Y. Won, F. S. Bates and D. E. Discher, Biotechnol. Bioeng., 2001, 73, 135–145 CrossRef CAS.
- W. Meier, C. Nardin and M. Winterhalter, Angew. Chem., Int. Ed., 2000, 39, 4599–4602 CrossRef.
- A. Taubert, A. Napoli and W. Meier, Curr. Opin. Chem. Biol., 2004, 8, 598–603 CrossRef CAS.
- J. Babin, J. Rodriguez-Hernandez, S. Lecommandoux, H.-A. Klok and M.-F. Achard, Faraday Discuss., 2005, 128, 179–192 RSC.
- F. Checot, S. Lecommandoux, Y. Gnanou and H.-A. Klok, Angew. Chem., Int. Ed., 2002, 41, 1339–1343 CrossRef CAS.
- K. E. Gebhardt, S. Ahn, G. Venkatachalam and D. A. Savin, J. Colloid Interface Sci., 2008, 317, 70–76 CrossRef CAS.
- H. Kukula, H. Schlaad, M. Antonietti and S. Foerster, J. Am. Chem. Soc., 2002, 124, 1658–1663 CrossRef CAS.
- R. Sigel, M. Losik and H. Schlaad, Langmuir, 2007, 23, 7196–7199 CrossRef CAS.
- H. R. Kricheldorf, Angew. Chem., Int. Ed., 2006, 45, 5752 CrossRef CAS.
- W. Agut, D. Taton and S. Lecommandoux, Macromolecules, 2007, 40, 5653–5661 CrossRef CAS.
- C. J. Hawker and K. L. Wooley, Science, 2005, 309, 1200–1205 CrossRef CAS.
- J.-F. Lutz, Angew. Chem., Int. Ed., 2007, 46, 1018–1025 CrossRef CAS.
- F. Sanda, G. Gao and T. Masuda, Macromol. Biosci., 2004, 4, 570–574 CrossRef CAS.
- B. S. Sumerlin and A. P. Vogt, Macromolecules, 2009, 43, 1–13.
- J. W. Chan, C. E. Hoyle and A. B. Lowe, J. Am. Chem. Soc., 2009, 131, 5751–5753 CrossRef CAS.
- A. Dondoni, Angew. Chem., Int. Ed., 2008, 47, 8995–8997 CrossRef CAS.
- C. E. Hoyle and C. H. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS.
- C. E. Hoyle, T. Y. Lee and T. Roper, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5301–5338 CrossRef CAS.
- J. Shin, H. Matsushima, J. W. Chan and C. E. Hoyle, Macromolecules, 2009, 42, 3294–3301 CrossRef CAS.
- B. D. Polizzotti, B. D. Fairbanks and K. S. Anseth, Biomacromolecules, 2008, 9, 1084–1087 CrossRef CAS.
- B. D. Fairbanks, M. P. Schwartz, A. E. Halevi, C. R. Nuttelman, C. N. Bowman and K. S. Anseth, Adv. Mater., 2009, 21, 5005–5010 CrossRef CAS.
- A. Aimetti, R. Shoemaker, C. Lin and K. Anseth, Chem. Commun., 2010, 46, 4061–4063 RSC.
- J. Sun and H. Schlaad, Macromolecules, 2010, 43, 4445–4448 CrossRef CAS.
- J. W. Chan, J. Shin, C. E. Hoyle, C. N. Bowman and A. B. Lowe, Macromolecules, 2010, 43, 4937–4942 CrossRef CAS.
- B. D. Fairbanks, T. F. Scott, C. J. Kloxin, K. S. Anseth and C. N. Bowman, Macromolecules, 2009, 42, 211–217 CrossRef CAS.
- R. M. Hensarling, V. A. Doughty, J. W. Chan and D. L. Patton, J. Am. Chem. Soc., 2009, 131, 14673–14675 CrossRef CAS.
- B. Yu, J. W. Chan, C. E. Hoyle and A. B. Lowe, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3544–3557 CrossRef CAS.
- M. Lo Conte, S. Pacifico, A. Chambery, A. Marra and A. Dondoni, J. Org. Chem., 2010, 75, 4644–4647 CrossRef CAS.
- A. Aimetti, K. Feaver and K. Anseth, Chem. Commun., 2010, 46, 5781–5783 RSC.
- J. Babin, D. Taton, M. Brinkmann and S. Lecommandoux, Macromolecules, 2008, 41, 1384–1392 CrossRef CAS.
- A. Gitsas, G. Floudas, M. Mondeshki, H. J. Butt, H. W. Spiess, H. Iatrou and N. Hadjichristidis, Biomacromolecules, 2008, 9, 1959–1966 CrossRef CAS.
- I. V. Dimitrov, I. V. Berlinova, P. V. Iliev and N. G. Vladimirov, Macromolecules, 2008, 41, 1045–1049 CrossRef CAS.
- B. D. Fairbanks, E. A. Sims, K. S. Anseth and C. N. Bowman, Macromolecules, 2010, 43, 4113–4119 CrossRef CAS.
- S. S. Naik and D. A. Savin, Macromolecules, 2009, 42, 7114–7121 CrossRef CAS.
Footnotes |
| † This paper is part of a Polymer Chemistry issue highlighting the work of emerging investigators in the polymer chemistry field. Guest Editors: Rachel O'Reilly and Andrew Dove. |
| ‡ Electronic supplementary information (ESI) available: Synthetic protocols, NMR and peak assignments, SLS, TEM, GPC and FTIR kinetic data. See DOI: 10.1039/c0py00231c |
| This journal is © The Royal Society of Chemistry 2011 |