Phosphatase/temperature responsive poly(2-isopropyl-2-oxazoline)†‡
Pier-Francesco
Caponi
a,
Xing-Ping
Qiu
b,
Filipe
Vilela
a,
Françoise M.
Winnik
b and
Rein V.
Ulijn
*a
aWestCHEM/Department of Pure and Applied Chemistry, The University of Strathclyde, Glasgow, G1 1XL, Scotland, UK. E-mail: rein.ulijn@strath.ac.uk; Fax: +44 (0)141 548 4822; Tel: +44 (0)141 548 2110
bDepartment of Chemistry and Faculty of Pharmacy, University of Montreal, Montreal, QC H3C 3J7, Canada
First published on 12th November 2010
Abstract
We demonstrate a strategy for producing polymer bioconjugates which display both enzymatic and thermal responsiveness. This is achieved by combining the thermo-responsive properties of poly(2-isopropyl-2-oxazoline) (PiPrOx) with the self-assembly properties of fluorenylmethoxycarbonyl-tyrosine (Fmoc-Y), controlled by a phosphatase triggered mechanism.
Significant attention has recently been focused on the development of so-called “smart” materials1 that can respond to external stimuli with a change in physical properties.2 These materials can be tailored to respond to various stimuli, e.g. temperature,3 ionic strength,4 pH5 or the catalytic activity of enzymes.6 Systems that respond to combined stimuli, such as pH and temperature,7 temperature and light,8 and temperature and enzymatic activity6d have also been described. Exploiting enzyme activity in this context has many advantages, including catalytic amplification of the self-assembly trigger, the ability to function under constant, physiological conditions and selectivity. Given the important role that enzymes play in biological pathways related to health and disease, this class of responsive materials may be expected to become increasingly important for future biomedical applications.1
Here, we focus on the use of phosphatase catalytic activity, combined with thermal responsiveness, to control polymer self-assembly. Phosphatases play a variety of vital roles in many organs in the human body where they are often expressed at cell surfaces, i.e. their catalytic sites are exposed and can interact with the cell's external environment. Their expression levels commonly vary significantly during processes of relevance to health, disease and repair, as is the case for bone regeneration.9 Moreover, their antagonistic disregulation with kinases is suspected to be involved in the development of cancer, diabetes and Alzheimer's syndrome.10
In 2004, Xu and co-workers6a,11a reported the first examples of phosphatase triggered molecular self-assembly, based on dephosphorylation of Fmoc-pY to form the self-assembling Fmoc-Y, thereby controlling changes in electrostatic interactions. A number of responsive systems have since been developed based on de-phosphorylation of peptide derivatives.9c,d More recently, similar switch systems have been introduced into polymeric materials, as demonstrated by Kühnle and Börner12 and Amir et al.13 These are elegant systems, but their response times were slow, at least in part because their kinetics are dictated by the enzymatic hydrolysis of multiple phosphate groups per polymer.
Here, we describe a system which combines the self-assembly properties of Fmoc-Y,6a with the thermal responsive properties of poly(2-isopropyl-2-oxazoline) (PiPrOx), thus achieving a dual responsiveness. In addition, we have developed a click chemistry route to polymer bioconjugation which will facilitate future development of similar hybrid polymeric/biomolecular systems with enzyme-responsiveness built-in.
A number of thermal responsive materials were reported in the last few years with poly(N-isopropylacrylamide) (PNIPAM) the most widely studied.14 Compared to PNIPAM, PiPrOx has a number of advantages. First, poly(2-alkyl-2-oxazoline) has low toxicity15—indeed, two members of this family, i.e. poly(2-methyl-2-oxazoline) and poly(2-ethyl-2-oxazoline), have obtained Food and Drug Administration (FDA) approval.16,17 Second, they show a so-called “stealth” behaviour,18 displaying reduced interactions with (immune system) proteins. Furthermore, the lower critical solution temperature (LCST) of PiPrOx (∼39 °C) is close to physiological temperatures and can be further tailored by modifying the polymer end groups.19
The synthesis of PiPrOx was performed by cationic ring opening polymerisation (CROP) of 2-isopropyl-2-oxazoline (Scheme 1a).20 Propargyl tosylate was used as the initiator in order to introduce clickable alkyne groups to the α-end of the polymer chains (for details on polymerisation and characterisation see ESI‡). Fmoc-pY was reacted with 11-azido-3,6,9-trioxaundecan-1-amine by the standard solid phase peptide synthesis procedure (see ESI‡) to obtain an Fmoc-pY-azide. Click chemistry was conducted in water using Cu(I) as catalyst generated in situ from the reduction of CuSO4 by sodium ascorbate (Scheme 1c).21 The level of end-functionalisation of the polymer was found to be 90% as evaluated by UV/Vis exploiting the absorbance of Fmoc at 300 nm.
 | ||
| Scheme 1 (a) CROP used to obtain propargyl-PiPrOx-OH (3). Propargyl toluene-4-sulfonate was used as initiator, while water was the terminating agent. (b) Coupling of Fmoc-pY with 11-azido-3,6,9-trioxaundecan-1-amine using a standard coupling protocol. (c) Click reaction used to obtain Fmoc-pY-PiPrOx-OH (1). | ||
Successful click coupling was indirectly evidenced by the changed phase transition temperature of aqueous propargyl-PiPrOx-OH before and after Fmoc-pY incorporation (Fig. 2). The LCST of 3 in water was 46 °C measured by UV transmittance, while 1 had a LCST value of 39.5 °C. This decrease in LCST is due to the presence of the hydrophobic Fmoc-pY functionality. The change in LCST could also be used to monitor enzyme triggered supramolecular rearrangements proposed in Fig. 1. When an aqueous solution containing 1 is left overnight in the presence of 50 U of phosphatase, the LCST decreased by 2 °C, due to the removal of hydrophilic phosphate groups, further enhancing the hydrophobicity (Fig. 2). The unmodified polymer showed no significant hysteresis as previously reported,22 hysteresis was observed for the functionalised polymers 1 and 2.
 | ||
| Fig. 1 (a) Dephosphorylation of Fmoc-pY-PiPrOx (1) catalysed by phosphatase. On average n = 48. (b) Schematic representation of enzyme- and temperature-induced self-assembly behaviour of the polymer bioconjugate. The phosphorylated polymer forms weak self-assembled structures due to the interaction between Fmoc-pY moieties (1a). Above the LCST, random aggregates are formed (1b). After cleavage of the phosphate groups, Fmoc-Y moieties drive the self-assembly into micelles (2a). Above the LCST, the corona structure of the micelles collapses around the Fmoc-Y core (2b). | ||
 | ||
| Fig. 2 Reversibility of phase transitions of Fmoc-pY-PiPrOx-OH (1) (●), Fmoc-Y-PiPrOx-OH (2) (▲) and propargyl-PiPrOx-OH (3) (■). Continuous line represents heating process while dotted line indicates cooling. | ||
The enzyme triggered self-assembly was also monitored by observing changes in fluorescence emission of fluorenes (Fig. 3). 50 U of enzyme were added to a 0.005 mg ml−1 solution of pY-Fmoc-PiPrOx-OH. 10 minutes after enzyme addition, a decrease of fluorescence intensity at 305 nm was noticeable and after 3 hours the fluorescence spectrum did not show further changes. In addition to peak at 305 nm, the emission spectra are characterised by relatively narrow peaks at approximately 320 nm and a broader peak at 370 nm. The latter is thought to represent fluorenyl excimers, indicative of Fmoc-Y aggregation. The ratio between the intensity of the 305 (monomer) and 370 (excimer) decreases with time (Fig. 3), suggesting enzyme action induces Fmoc-Y aggregation.
 | ||
| Fig. 3 Enzymatic triggered self-assembly monitored by fluorescence spectroscopy. The inset graph shows the decrease in fluorescence intensity after addition of the enzyme over the course of 3 hour reaction (increasing time from top to bottom). | ||
To confirm the reversibility of the thermal response process, several cycles of heating and cooling (above and below the LCST) were completed for the polymer before (1) and after enzymatic (2) treatment. After several cycles, both the polymers (1 and 2) are capable to revert back to the initial soluble state (see ESI‡).
Finally, dynamic light scattering (DLS) was used to confirm that nano-objects with defined sizes form upon enzyme action. The average hydrodynamic radius of 3 in aqueous solution at room temperature was ∼2 nm (Fig. 4), while the Rh values of 1 and 2 were 44 and 92 nm, respectively. These observations indicate that before functionalization, the polymer 3 does not aggregate, while after functionalisation, micellar structures form (Fig. 1), with their sizes dictated by the presence of phosphate groups. Above the LCST, the polymer obtained after enzymatic treatment forms small mesoglobules (Rh = 21 nm) as a result of the dehydration collapse and aggregation of the poly(2-isopropyl-2-oxazoline) chains (Fig. 1 and 5).
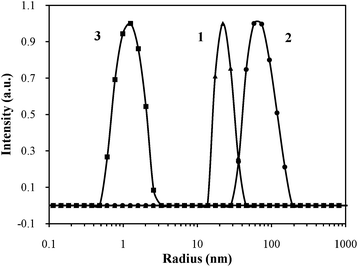 | ||
| Fig. 4 DLS data showing the average particle size dimension at room temperature being 44 nm for Fmoc-pY-PiPrOx-OH (1) (▲), 92 nm for Fmoc-Y-PiPrOx-OH (2) (●) and 2 nm for propargyl-PiPrOx-OH (3) (■). | ||
 | ||
| Fig. 5 Hydrodynamic diameter of Fmoc-pY-PiPrOx-OH (1) (■), Fmoc-Y-PiPrOx-OH (2) (▲) and propargyl-PiPrOx-OH (3) (●) as a function of the solution temperature (polymer concentration = 2.5 mg ml−1). | ||
To fully understand the nature of structures that are formed by 1 and 2 further studies will be required. We hypothesise that 1 randomly collapses above LCST, while, upon enzymatic cleavage, aromatic interactions of Fmoc-Y6a give rise to shell–corona-like structures that above LCST form stable micelles.
The system presented here is a first example of phosphatase/temperature dual responsive polymers. Starting from two well-known building blocks for stimuli-responsive materials we have been able to create a novel responsive material using a straightforward synthetic methodology which can be expanded to different responsive polymer/enzyme substrates. We are currently developing these materials further to release bioactive payloads in response to cell surface phosphatases, in an effort to control and direct cellular behaviour.23
The authors thank WestCHEM and JCE MolChem program and EPSRC (Advanced Research Fellowship RVU) for funding, Patricia Keating for MALDI and Katarzyna Sypek for helping with DLS experiment.
Notes and references
- J. S. Mohammed and W. L. Murphy, Adv. Mater., 2009, 21, 2361–2374 CrossRef CAS.
- (a) R. V. Ulijn, J. Mater. Chem., 2006, 16, 2217–2225 RSC; (b) Z. Yang, G. Liang and B. Xu, Acc. Chem. Res., 2008, 41, 315–326 CrossRef CAS; (c) H. G. Börner, H. Kühnle and J. Hentschel, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1–14 CrossRef CAS.
- A. S. Hoffman and P. Stayton, Prog. Polym. Sci., 2007, 32, 922–932 CrossRef CAS.
- G. A. Silva, C. Czeisler, K. L. Niece, E. Beniash, D. A. Harrington, J. A. Kessler and S. L. Stupp, Science, 2004, 303, 1352–1355 CrossRef CAS.
- (a) C. Tang, A. M. Smith, R. F. Collins, R. V. Ulijn and A. Saiani, Langmuir, 2009, 25, 9447–9453 CrossRef CAS; (b) L. Chen, S. Revel, K. Morris, L. C. Serpell and D. J. Adams, Langmuir, 2010, 26, 13466–13471 CrossRef CAS.
- (a) Z. M. Yang, H. W. Gu, D. G. Fu, P. Gao, J. K. Lam and B. Xu, Adv. Mater., 2004, 16, 1440–1444 CrossRef; (b) P. D. Thornton, R. J. Mart, S. Webb and R. V. Ulijn, Soft Matter, 2008, 4, 821–827 RSC; (c) P. D. Thornton, R. J. Mart and R. V. Ulijn, Adv. Mater., 2007, 19, 1352–1256 CrossRef CAS; (d) K. J. C. Van Bommel, M. C. A. Stuart, B. L. Feringa and J. Van Esch, Org. Biomol. Chem., 2005, 3, 2917–2920 RSC; (e) A. R. Hirst, S. Roy, M. Arora, A. K. Das, N. Hodson, P. Murray, S. Marshall, N. Javid, J. Sefcik, J. Boekhoven, J. Van Esch, S. Santabarbara, N. T. Hunt and R. V. Ulijn, Nat. Chem., 2010, 2 DOI:10.1038/NCHEM.861.
- (a) D. Roy, J. N. Cambre and B. S. Sumerlin, Prog. Polym. Sci., 2010, 35, 278–301 CrossRef CAS; (b) N. Suchao-In, S. Chirachanchai and S. Perrier, Polymer, 2009, 50, 4151–4158 CrossRef CAS.
- S. Dai, P. Ravi and K. C. Tam, Soft Matter, 2009, 5, 2513–2533 RSC.
- E. E. Golub and K. Boesze-Battaglia, Curr. Opin. Orthop., 2007, 18, 444–448 CrossRef.
- J. N. Andersen, P. G. Jansen, S. M. Echwald, O. H. Mortensen, T. Fukada, R. Del Vecchio, N. K. Tonks and N. P. H. Møller, FASEB J., 2004, 18, 8–30 CrossRef CAS.
- (a) Z. Yang and B. Xu, Chem. Commun., 2004, 2424–2425 RSC; (b) Y. Gao, Y. Kuang, Z. F. Guo, Z. H. Guo, I. J. Krauss and B. Xu, J. Am. Chem. Soc., 2009, 131, 13576–13577 CrossRef CAS; (c) W. Wang and Y. Chau, Soft Matter, 2009, 5, 4893–4898 RSC.
- H. Kühnle and H. G. Börner, Angew. Chem., Int. Ed., 2009, 48, 6431–6434 CrossRef.
- J. A. Amir, S. Zhong, D. J. Pochan and C. J. Hawker, J. Am. Chem. Soc., 2009, 131, 13949–13951 CrossRef CAS.
- (a) D. Schmaljohann, Adv. Drug Delivery Rev., 2006, 58, 1655–1670 CrossRef CAS; (b) J. F. Mano, Adv. Eng. Mater., 2008, 10, 515–527 CrossRef CAS.
- P. Goddard, L. E. Hutchinson, J. Brown and L. J. Brookman, J. Controlled Release, 1989, 10, 5–16 CrossRef CAS.
- M. C. Woodle, C. M. Engbers and S. Zalipsky, Bioconjugate Chem., 1994, 5, 493–496 CrossRef CAS.
- S. Zalipsky, C. B. Hansen, J. M. Oaks and T. M. Allen, J. Pharm. Sci., 1996, 85, 133–137 CrossRef CAS.
- R. Hoogenboom, Angew. Chem., Int. Ed., 2009, 48, 7978–7994 CrossRef CAS.
- S. Huber, N. Hutter and R. Jordan, Colloid Polym. Sci., 2008, 286, 1653–1661 CrossRef CAS.
- R. Hoogenboom, F. Wiesbrock, H. Huang, M. A. A. Leenen, H. M. L. Thijs, S. F. G. M. van Nispen, M. van der Loop, C. A. Fustin, A. M. Jonas, J. F. Gohy and U. S. Schubert, Macromolecules, 2006, 39, 4719–4725 CrossRef CAS.
- R. Luxenhofer and R. Jordan, Macromolecules, 2006, 39, 3509–3516 CrossRef CAS.
- R. Hoogenboom, H. M. L. Thijs, M. J. H. C. Jochems, B. M. van Lankvelt, M. W. M. Fijten and U. S. Schubert, Chem. Commun., 2008, 5758–5760 RSC.
- J. H. Gao and B. Xu, Nano Today, 2009, 4, 37–51 CrossRef.
Footnotes |
| † This paper is part of a Polymer Chemistry issue highlighting the work of emerging investigators in the polymer chemistry field. Guest editors: Rachel O'Reilly and Andrew Dove. |
| ‡ Electronic supplementary information (ESI) available: Experimental procedures, polymer synthesis, polymer characterisation data, Fmoc-pY-trioxaundecan-azide synthesis and characterisation, thermal cycles. See DOI: 10.1039/c0py00291g |
| This journal is © The Royal Society of Chemistry 2011 |