Thermoresponsive giant biohybrid amphiphiles†‡
Christine
Lavigueur
a,
Jordi González
García
a,
Linda
Hendriks
a,
Richard
Hoogenboom
a,
Jeroen J. L. M.
Cornelissen
*b and
Roeland J. M.
Nolte
a
aRadboud University Nijmegen, Institute for Molecules and Materials, Department of Organic Chemistry, P.O. Box 9010, 6500 GL, Nijmegen, The Netherlands
bLaboratory for Biomolecular Nanotechnology, MESA+ Institute for Nanotechnology, University of Twente, P.O. Box 217, 7500 AE, Enschede, The Netherlands. E-mail: j.j.l.m.cornelissen@tnw.utwente.nl; Fax: +31 53 489 4645; Tel: +31 53 489 4380
First published on 22nd September 2010
Abstract
A series of random copolymers of various lengths was prepared by atom transfer radical polymerisation (ATRP) using two acrylate monomers with short pendant ethylene glycol side chains (ethylene glycol methyl ether acrylate, EGMEA, and methoxy ethoxy ethyl acrylate, MEEA). The end group was converted to an azide to enable bioconjugation through copper-catalysed azide–alkyne cycloaddition (CuAAC). All polymers were found to be thermoresponsive, with a cloud point between 25 and 35 °C depending on their molecular weight. They were conjugated to enhanced green fluorescent protein (EGFP) functionalised with a single alkyne moiety, as seen by fast performance liquid chromatography (FPLC) and gel electrophoresis (SDS-PAGE). The resulting biohybrid amphiphiles were thermoresponsive. Dynamic light scattering (DLS) and transmission electron microscopy (TEM) were used to study their self-assembly at elevated temperatures, and they were found to form spherical structures with a diameter of approximately 60 nm upon slow heating.
Introduction
Amphiphilic diblock copolymers, consisting of a hydrophobic and a hydrophilic segment, have been shown to form self-assembled structures in aqueous media with a wide range of morphologies.1–4 These structures have been proposed for a variety of applications, ranging from drug-delivery to nanoreactors.5,6 More recently, a class of larger amphiphiles has been reported, the giant amphiphiles, which are formed by linking a hydrophobic polymer tail to a hydrophilic protein head.7–10 These giant biohybrid amphiphiles were also shown to self-assemble in aqueous media, where they can form biologically relevant self-assembled aggregates that inherently incorporate the desired protein or enzyme. Several examples of giant amphiphiles have been reported,11–18 most commonly using polystyrene as the polymer tail.12–18 Because of this choice of polymer, the two parts of the amphiphiles do not share any common solvent, which implies that the self-assembly cannot be observed at equilibrium and that any obtained structures will be kinetically trapped.In order to better understand the self-assembly process of giant amphiphiles, we are interested in designing a system that would enable the study of their self-assembly at equilibrium. A promising avenue is to design thermoresponsive giant amphiphiles, which can self-assemble upon slow heating.
A wide variety of thermoresponsive polymers, which undergo a reversible transition from a water-soluble to a water-insoluble state upon heating above a critical temperature (lower critical solution temperature, LCST), have been reported.19–21 The most commonly studied among these so-called “smart” polymers is poly(N-isopropylacrylamide) (PNIPAM).22–24 The popularity of this polymer can be explained by its sharp transition, tuneable LCST near body temperature, and relative insensitivity to environmental conditions. It does, however, present important drawbacks such as suspected toxicity, irreversibility of the transition (significant hysteresis), and the presence of amides which can lead to potentially problematic hydrogen bonding interactions with proteins.
Acrylate or methacrylate polymers with short oligoethylene glycol (OEG) side chains have been shown to possess thermoresponsive properties comparable to those of PNIPAM, but without the most important drawbacks.25–28 Notably, since these polymers are mostly composed of OEG, they are generally fully biocompatible. By varying the length of the OEG side chain or by changing the ratio of monomers having different side chain lengths, it is possible to adjust the transition temperature of these polymers.27–29Methacrylate based OEG polymers are more common, but acrylate polymers have also been reported.27,30–32Acrylate polymers tend do be easier to end-functionalise than methacrylate polymers,33 they are therefore promising candidates for bioconjugation with a grafting-to approach.
Thermoresponsive polymers have previously been coupled to a variety of proteins to produce temperature-responsive protein–polymer conjugates,34,35 PNIPAM being by far the most commonly used thermoresponsive polymer for bioconjugation.36–44 In many examples, several thermoresponsive polymer chains were coupled to each protein,40,42,43 often with the aim to precipitate the protein at elevated temperature as a purification strategy. It was also reported that a single polymer chain attached to a protein is sufficient to confer thermoresponsive properties to the conjugate.36–39 However, the self-assembly of giant thermoresponsive amphiphiles has not been investigated in details to date.
In this article, we describe the preparation and self-assembly of thermoresponsive giant biohybrid amphiphiles derived from random copolymers of ethylene glycol methyl ether acrylate (EGMEA) and methoxy ethoxy ethyl acrylate (MEEA), P(EGMEA,MEEA)n, which were conjugated to enhanced green fluorescent protein (EGFP). The novel azide-terminated thermoresponsive polymers were prepared through atom transfer radical polymerisation (ATRP), while an alkyne functionality was incorporated to the model protein, EGFP, at a naturally occurring cysteine. Their bioconjugation through copper-catalysed azide–alkyne cycloaddition (CuAAC) and the study of the temperature dependent self-assembly of the obtained thermoresponsive giant biohybrid amphiphiles are reported.
Results and discussion
Random copolymers of EGMEA and MEEA, P(EGMEA,MEEA)n, were prepared by ATRP using ethyl 2-bromoisobutyrate (EBiB) as the initiator, and copper bromide and pentamethyldiethylenetriamine (PMDETA) as the catalyst, as outlined in Scheme 1. The polymerisations were monitored by 1H NMR and stopped at ∼45 to 70% conversion. A linear relationship was observed between ln([M0]/[Mt]) and reaction time, as seen in Fig. 1, suggesting good control of the polymerisations. Furthermore, gel permeation chromatography (GPC) indicated that the polydispersity indices (PDI) of all polymers were below 1.15. Variations in the reaction kinetics were likely caused by changes in the scale of the reactions and in the amount of anisole used in the different polymerisations. The bromine end group was converted to an azide using sodium azide. | ||
| Scheme 1 Synthesis of the thermoresponsive random copolymers P(EGMEA,MEEA)n by ATRP. EBiB: ethyl 2-bromoisobutyrate; PMDETA: pentamethyldiethylenetriamine; and DMF: dimethylformamide. | ||
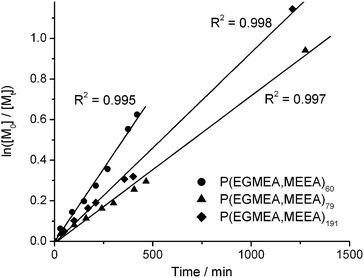 | ||
| Fig. 1 Kinetic plot of the ATRP of EGMEA and MEEA at 75 °C showing first order kinetics, as monitored by 1H NMR spectroscopy. | ||
Table 1 lists the reaction conditions and characteristics of the polymers used in this study. Mass spectrometry and 1H NMR data showed that the composition of the polymers reflects the monomer feeds, with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of EGMEA and MEEA.‡ The thermoresponsive behaviour of the azide-terminated polymers was studied by turbidimetry. As seen in Fig. 2, all polymers were found to exhibit a cloud point between 25 and 35 °C, the temperature of the transition decreasing with increasing molecular weight. Interestingly, dependence of the transition temperature on molecular weight was also observed with similar acrylate polymers having short OEG side chains,32 but not for related methacrylate polymers incorporating longer side chains.28 The polymers could reversibly undergo many heating and cooling cycles.‡ It was also observed that the cloud point shifts to higher temperatures when the polymer concentration is decreased, or when the concentration of added salt is decreased.‡
:1 ratio of EGMEA and MEEA.‡ The thermoresponsive behaviour of the azide-terminated polymers was studied by turbidimetry. As seen in Fig. 2, all polymers were found to exhibit a cloud point between 25 and 35 °C, the temperature of the transition decreasing with increasing molecular weight. Interestingly, dependence of the transition temperature on molecular weight was also observed with similar acrylate polymers having short OEG side chains,32 but not for related methacrylate polymers incorporating longer side chains.28 The polymers could reversibly undergo many heating and cooling cycles.‡ It was also observed that the cloud point shifts to higher temperatures when the polymer concentration is decreased, or when the concentration of added salt is decreased.‡
:MEEA:EBiB. Polymer properties are for the final azide-terminated polymers
| Ratio | Conv.a (%) | n b | M MALDI b/g mol−1 | M nGPC c/g mol−1 | PDIc |
|---|---|---|---|---|---|
| a The % conversion was determined by 1H NMR spectroscopy monitoring of the reaction. b The degree of polymerisation (n) and molecular weight (MMALDI) were evaluated by MALDI-TOF MS. c The number average molecular weight (MnGPC) and PDI were measured by GPC using a polystyrene calibration. | |||||
| 100:100:1 |
46 | 60 | 9433 | 8271 | 1.08 |
| 100:100:1 |
61 | 79 | 12185 |
10569 |
1.13 |
| 220:220:1 |
68 | 191 | 29268 |
26684 |
1.13 |
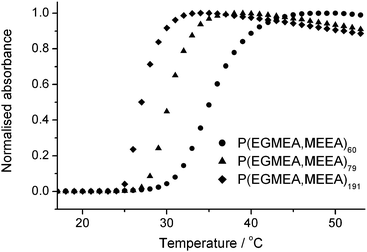 | ||
| Fig. 2 Cloud point profile of the three random copolymers measured on 0.5% (w/v) solutions in phosphate buffered saline (PBS, pH 8.0, 50 mM, 0.1 M NaCl) at a scanning rate of 1 °C min−1. | ||
In order to prepare well-defined biohybrid amphiphiles, it is desirable to attach a single polymer tail to a protein head group. EGFP has two cysteine residues that could potentially offer an available thiol, as depicted in Scheme 2. However, one of these cysteines is located inside the β-barrel, and is not expected to be available for functionalisation. Ellman's assay confirmed that less than one thiol is available per protein. EGFP was coupled to N-propargyl maleimide to introduce an alkyne functionality on which the polymer could be attached through CuAAC, as detailed in Scheme 2. Mass spectrometry measurements indicated that a single maleimide was successfully attached to the protein. It was also noted that the EGFP used had slightly degraded, loosing residues at the C-terminus.‡
 | ||
| Scheme 2 Synthesis of the biohybrids and schematic representation of the position of the two cysteine groups on EGFP (structure 1GFL from the PDB, the His-tag and linker are not included). PBS: phosphate buffered saline; THF: tetrahydrofuran; and MeCN: acetonitrile. | ||
The conjugation strategy was first tested using the profluorescent dye 3-azido-7-hydroxy coumarin, which is known to become fluorescent only after having undergone CuAAC.45,46Alkyne-functionalised EGFP was coupled to the dye, following which the catalyst and most of the unbound coumarin were removed by dialysis. The protein used had a His-tag at the N-terminus, which allowed it to bind to Ni-NTA agarose beads; this technique was employed to wash away any residual free coumarin remaining after dialysis. An intense absorption band corresponding to the coumarin dye was visible by UV-visible spectroscopy, as shown in Fig. 3a. A fluorescence band centred around λ = 470 nm was also observed when exciting at λ = 380 nm, as seen in Fig. 3b, which is indicative that the profluorescent dye has been conjugated to the alkyne through the CuAAC reaction. When a control reaction was performed without catalyst, almost no coumarin absorption or emission was observed.
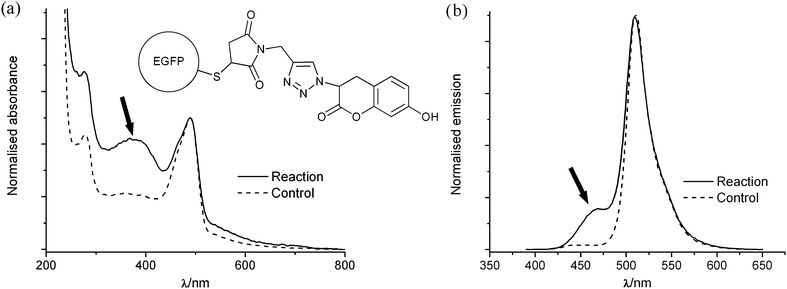 | ||
| Fig. 3 (a) UV-visible spectra and (b) fluorescence spectra (excitation λ = 380 nm) of the product of a test reaction between acetylene-functionalised EGFP and 3-azido-7-hydroxy coumarin performed either with the catalyst present (reaction) or without (control). The spectra were normalised to have the same absorbance or emission at the maxima of the EGFP peak; the coumarin bands are indicated with arrows. | ||
The polymers were conjugated to EGFP in the same manner as was used for the coumarin dye. The catalyst and ligand were removed by dialysis, following which the biohybrids were bound to Ni-NTA agarose beads and the excess polymer was washed away. For each sample, fast performance liquid chromatography (FPLC) showed the presence of unreacted EGFP and of a larger species consistent with the formation of the desired biohybrid, as seen in Fig. 4. As the length of the polymer tail was increased, the peak corresponding to the biohybrid shifted to lower elution volumes. In the case of the biohybrid incorporating the longest polymer chain, this peak was quite well separated from the peak of EGFP, whereas when the shortest polymer chain was used, only a shoulder was observed on the side of the EGFP peak. The fractions corresponding to the desired product were isolated in order to remove a large portion of the unreacted protein.
 | ||
| Fig. 4 FPLC trace of EGFP and of the three biohybrids ran on a Superdex 75 column and monitored at λ = 490 nm. | ||
The obtained biohybrids were also analysed by gel electrophoresis (SDS-PAGE), as shown in Fig. 5. Unmodified EGFP and a larger species were both observed in the crude samples, while samples purified by FPLC contained much less of the starting protein, confirming that isolation by FPLC successfully removed most of the unreacted protein. This was also confirmed by re-analysing the purified samples by FPLC.‡
 | ||
| Fig. 5 SDS-PAGE of the crude and purified biohybrids. Lane 1: EGFP; lanes 2, 3 and 4: biohybrids after Ni-NTA column treatment EGFP–P(EGMEA,MEEA)nn = 60, 79, and 191 respectively; lanes 5, 6, and 7: the biohybrids after FPLC purification EGFP P(EGMEA,MEEA)nn = 60, 79, and 191 respectively. | ||
The thermoresponsive behaviour of the giant biohybrid amphiphiles was monitored by dynamic light scattering (DLS). The derived count rate (scattering intensity) was measured at 1 °C intervals while heating from 15 to 55 °C; results are shown in Fig. 6. The biohybrid having the longest polymer tail, EGFP–P(EGMEA,MEEA)191, showed a clear transition centred at 35 °C, while EGFP–P(EGMEA,MEEA)79 exhibited a broader transition centred around 45 °C. The biohybrid having the shortest polymer tail, EGFP–P(EGMEA,MEEA)60, displayed a slight increase in turbidity upon heating, but no clear transition. It is likely that the transition was not completed at the end temperature of the measurement and that the turbidity would have risen more sharply upon further heating. This was, however, not possible with the experimental setup used and could have eventually led to degradation of the protein. Crucially, the results from EGFP–P(EGMEA,MEEA)191 indicate that while the number of aggregates formed increases over the course of the transition, thus increasing the turbidity, their size does not vary significantly. It is therefore appropriate to examine the size of the self-assembled structures formed by EGFP–P(EGMEA,MEEA)60 at 55 °C even if the transition is not completed at this temperature.
 | ||
| Fig. 6 Cloud point diagram of EGFP and the biohybrids monitored by DLS (for experimental details see Experimental section). | ||
The size of the particles present in solution at low or high temperature was also monitored by DLS, as exemplified in Fig. 7 and detailed in Table 2. At 5 or 10 °C, all three giant amphiphiles had a hydrodynamic diameter (Dh) only slightly larger than that of EGFP at a similar temperature. Upon heating the biohybrids to 55 °C, the peak corresponding to single amphiphiles disappeared and larger aggregates were observed. These aggregates were found to have a similar size for all three amphiphiles, although a small increase in diameter was observed with increasing length of the polymer tail. In contrast, EGFP did not show a pronounced size difference upon heating, clearly indicating that the bioconjugated polymers are responsible for the thermoresponsive behaviour of the biohybrids.
 | ||
| Fig. 7 Examples of volume size distribution obtained by DLS for EGFP and the biohybrids at (a) 5 °C (except EGFP–P(EGMEA,MEEA)60 shown at 10 °C) and (b) 55 °C (for experimental details see Experimental section). | ||
| T/°C | EGFP | EGFP–P(EGMEA,MEEA)60 | EGFP–P(EGMEA,MEEA)79 | EGFP–P(EGMEA,MEEA)191 | ||||
|---|---|---|---|---|---|---|---|---|
| 5 | 55 | 10 | 55 | 5 | 55 | 5 | 55 | |
| a Average hydrodynamic diameter (Dh) is from the DLS intensity size distribution. Low temperature values are averages of 6 runs and high temperature values are averages of 18 runs. b Average diameters (DTEM) are from TEM micrographs; between 160 and 260 particles were measured for each amphiphile. | ||||||||
| Avg. Dha | 3.6 | 6 | 9.1 | 63 | 10.5 | 64 | 8.0 | 71 |
| St. dev. | 0.2 | 1 | 0.6 | 9 | 0.3 | 7 | 0.4 | 5 |
| Avg DTEMb | — | — | — | 42 | — | 61 | — | 62 |
| St. dev. | — | — | — | 7 | — | 11 | — | 15 |
Transmission electron microscopy (TEM) was used in order to probe the morphology of the aggregates formed by the thermoresponsive amphiphiles at high temperature. Solutions of the biohybrids were slowly heated and left to equilibrate before being deposited on heated grids. Roughly circular structures were observed for all three samples, although they were not always perfectly well defined. This could be caused in part by the low glass transition temperature of these types of polymers. Size distributions and sample pictures are shown in Fig. 8 and average diameters (DTEM) are listed in Table 2.
 | ||
| Fig. 8 Size distribution measured from TEM micrographs and example of micrographs for (a) EGFP–P(EGMEA,MEEA)60; (b) EGFP–P(EGMEA,MEEA)79; and (c) EGFP–P(EGMEA,MEEA)191. Scale bars represent 200 nm. | ||
As expected, the sizes measured by TEM (dry diameter) were slightly smaller than those measured by DLS (hydrodynamic diameter). Furthermore, the sizes measured by TEM for all three amphiphiles were very similar but showed a slight increase with increasing polymer length, as had been observed by DLS. The shape and size of the observed aggregates would be approximately consistent with the formation of self-assembled micelles. Apparently, the size of the polymers is too small in relation to the EGFP to induce other types of micellar structures.
Conclusions
A series of well-defined giant amphiphiles were successfully prepared and were found to be thermoresponsive. Slow heating resulted in the formation of spherical aggregates roughly consistent with the expected size and shape of self-assembled micelles. Increasing the length of the polymer tail led to a slight increase in the size of the obtained aggregates. It is expected that a greater increase in the length of the polymer tail or a change in the shape of the protein head could lead to the formation of self-assembled structures having different morphologies. Further studies are currently under way.Experimental
Materials
Diethyl ether, n-heptane, DMF and acetonitrile were purchased from J. T. Baker. Dichloromethane (DCM) was purchased from Fisher Scientific. THF CHROMASOLV for HPLC was purchased from Sigma Aldrich. Deuterated chloroform was purchased from Cambridge Isotope Laboratories, Inc. All solvents were used as received. Deionised water with a resistivity of 18.2 MΩ cm−1 was obtained with a Labconco Water Pro PS purification system and was used for all experiments.PMDETA, anisole, ethylenediaminetetraacetic acid (EDTA), sodium azide and sodium chloride were purchased from Acros Organics. EBiB, 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB), sodium ascorbate, dithranol and imidazole were purchased from Sigma-Aldrich. Copper sulfate pentahydrate, Na2HPO4·2H2O and NaH2PO4·H2O were purchased from Merk. Uranyl acetate dihydrate was purchased from Fluka. Aluminium oxide, activated, neutral, 50–200 micron was purchased from Acros Organics. Ni-NTA agarose beads were purchased from Quiagen.
EGMEA was purchased from Acros Organics and MEEA was purchased from ABCR. The monomers were filtered through a short column of neutral alumina to remove the stabilisers immediately before carrying out each polymerisation. Copper bromide was purchased from Aldrich. It was stirred overnight in glacial acetic acid, filtered and washed with methanol, dried under vacuum, then stored under argon.
N-Propargyl maleimide,473-azido-7-hydroxy coumarin,46 and tris((1-((O-ethyl)carboxymethyl)-(1,2,3-triazol-4-yl))methyl)amine48,49 were prepared following literature procedures. EGFP was expressed in Escherichia coli, following published protocols.50
Instrumentation
1H NMR and 13C NMR spectra were recorded on a Bruker DPX300 spectrometer. FT-IR spectra were measured on an ATI Matson Genesis Series FT-IR spectrometer with a fitted ATR cell. UV-vis spectra were recorded on a Varian Cary 50 spectrophotometer using a quartz cuvette. Fluorescence spectra were acquired using a Perkin-Elmer luminescence spectrometer LS 55.Matrix assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) was carried out on a Bruker Biflex III spectrometer. Samples were prepared by mixing a THF solution of the polymer (1 µL, 7 mg mL−1) with a THF solution of the matrix, dithranol (10 µL, 20 mg mL−1). Electrospray ionization time-of-flight mass spectrometry (ESI-TOF) was carried out on a JEOL AccuTOF spectrometer.
Protein samples (5–10 µM protein) were prepared as solutions in deionised water or in dilute formic acid (0.1–1% (v/v) formic acid in deionised water). Deconvoluted spectra were obtained using the Magtran software.
Gel permeation chromatography (GPC) was carried out on a Shimadzu SEC equipped with a guard column and a PL gel 5 µm mixed D column from Polymer Laboratories, and with differential refractive index (RI) detection. THF was used as the eluent (1 mL min−1 at 35 °C) and polystyrene standards were used for the calibration. Turbidity studies were performed on a Jasco J-810 spectropolarimeter equipped with a Jasco PFD-425S temperature control unit, using a quartz cuvette.
FPLC was performed on an Amersham Biosciences Ettan LC system fitted with a Frac-950 fraction collector using a Superdex 75 PC 3.2/30 column from Pharmacia LKB Biotechnology. Buffers for FPLC were filtered with a Millipore 0.2 µM filter before use. Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) was performed using 12% (w/v) polyacrylamide gels, followed by silver staining.
Dynamic light scattering measurements were carried out on a Malvern Instrument Zetasizer Nano-S (ZEN1600) equipped with a He–Ne laser (633 nm, 4 mW) and an Avalanche photodiode detector at an angle of 173°. Transmission electron microscopy (TEM) was performed on a JEOL 1010 microscope at an acceleration voltage of 60 kV. Samples were deposited on carbon-coated Cu grids (200 mesh).
ATRP polymerisations of P(EGMEA,MEEA)n
Purified CuBr (1 eq.) was put in a Schlenk tube and placed under argon atmosphere. Purified EGMEA and MEEA (1:1 molar ratio, total weight 3.5–4.7 g, number of equivalents listed in Table 1 for each polymerisation) were added along with PMDETA (1 eq.) and anisole as internal standard and co-solvent (25–50 eq.). The mixture was degassed by two freeze–pump–thaw cycles. EBiB (1 eq.) was added along with a second portion of anisole (25–50 eq.) and the reaction mixture was degassed through one more freeze–pump–thaw cycle before being put under argon and placed in an oil bath at 75 °C with stirring. The reaction was monitored by 1H NMR spectroscopy until the desired conversion was reached (between 45 and 70%), after which it was placed in an ice bath, exposed to air, and diluted with DCM (40 mL).
The reaction mixture was extracted with aqueous EDTA (2 × 50 mL) and water (2 × 50 mL) to remove the catalyst. The organic layer was evaporated to dryness under reduced pressure. The product was purified by two precipitations from minimal DCM into cold heptane:ether (1:1, v/v) to obtain the product as a very viscous oil.
Azide-terminated P(EGMEA,MEEA)n
P(EGMEA,MEEA)n (∼1.2 g), sodium azide (40 eq.) and DMF (30 mL) were placed in a round bottom flask and stirred overnight at room temperature and under ambient atmosphere. The reaction mixture was evaporated to dryness under reduced pressure. The product was dissolved in DCM and filtered to remove the sodium azide, then evaporated to dryness under reduced pressure. The polymer was purified by precipitation from minimal DCM into cold heptane:ether (1:1, v/v) to obtain the product as a very viscous oil.
Polymer turbidity studies
Solutions of 0.5% (w/v) were prepared in PBS (pH 8.0, 50 mM, 0.1 M NaCl). Measurements were taken using a 1 cm path length cuvette. Samples were heated at 1 °C min−1 and the absorbance at 600 nm was measured at every 1 °C.Ellman's assay
A solution of EGFP (0.05 mM) and a solution of 5,5′-dithiobis-(2-nitrobenzoic acid), DTNB, (10 mM) were prepared in PBS (pH 8.0, 50 mM, 0.5 M NaCl). An aliquot of 25 µL of the DTNB solution was added to 500 µL of PBS; the UV-vis absorbance spectra of this solution and of the EGFP solution were measured. An aliquot of 25 µL of DTNB solution was added to 500 µL of EGFP solution and a spectrum was recorded every 5 minutes, until no further changes were observed after a total of 15 minutes.The absorbance of the protein solution and of the DTNB solution was subtracted from the absorbance of the EGFP–DTNB measured after 15 minutes of equilibration time. To estimate the number of free SH present, an extinction coefficient of 13 650 M−1 cm−1 at 410 nm was used for the protein–DTNB complex, giving a result of less than one equivalent SH per EGFP.
Alkyne-functionalised EGFP
N-Propargyl maleimide (4.7 mg, 0.035 mmol, 200 eq.) was dissolved in THF (100 µL). A solution of EGFP (0.13 mM, 1.33 mL, 0.18 µmol) in PBS (pH 7.2, 20 mM, 0.1 M NaCl) was added and the solution was stirred gently in the dark at 4 °C for 2 hours. The mixture was dialysed against PBS (pH 7.2, 20 mM, 0.1 M NaCl) to remove the excess maleimide and THF, then against PBS (pH 8.0, 50 mM, 0.1 M NaCl).Preparation of “click-mix” catalyst solution
The CuAAC reactions were performed using the following catalyst solution (click-mix), adding 1 µL of click-mix per nmol of protein (50 eq. copper sulfate; 63 eq. sodium ascorbate; and 98 eq. ligand). The tristriazole ligand described by Davis et al.49tris((1-((O-ethyl)carboxymethyl)-(1,2,3-triazol-4-yl))methyl)amine was dissolved in acetonitrile (2 mg µL−1). Copper sulfate pentahydrate and sodium ascorbate were dissolved in deionised water (5 mg mL−1 each), and this solution was used within a few minutes. The two solutions were mixed in a 1:1 (v/v) ratio to obtain the final click-mix, which was immediately added to the desired reaction mixture.
EGFP coumarin conjugation
A solution of alkyne-functionalised EGFP in PBS (0.17 mM, 90 µL, 0.015 µmol) was added to a solution of 3-azido-7-hydroxy coumarin (1.6 mM, 190 µL, 0.3 µmol, 20 eq.) in PBS (pH 8.0, 50 mM, 0.1 M NaCl). Fresh click-mix was added (73 µL) and the solution was stirred gently in the dark at 4 °C overnight.The mixture was dialysed against PBS (pH 8.0, 50 mM, 0.3 M NaCl) to remove the catalyst solution, then incubated with Ni-NTA agarose beads (from 90 µL of slurry) at 4 °C for 4 hours. The flow-through was collected and the column was washed (2 mL, PBS pH 8.0, 50 mM, 0.3 M NaCl, 12.5 mM imidazole) to remove the excess dye. The product was eluted from the column (∼0.6 mL, PBS pH 8.0, 50 mM, 0.3 M NaCl, 250 mM imidazole), dialysed against PBS (pH 8.0, 50 mM, 0.1 M NaCl), and concentrated to 100 µL.
The control reaction was performed in the same manner, but adding a deionised water:acetonitrile (1:1, v/v) mixture instead of the click-mix.
Preparation of the biohybrid amphiphiles
A solution of alkyne-functionalised EGFP (0.16 mM, 280 µL, 0.044 µmol) in PBS was added to a solution of the desired polymer (1.6 mM, 560 µL, 0.88 µmol, 20 eq.) in PBS (pH 8.0, 50 mM, 0.1 M NaCl). Fresh click-mix was added (219 µL) and the solution was gently stirred in the dark at 4 °C overnight.The mixture was dialysed against PBS (pH 8.0, 50 mM, 0.3 M NaCl) to remove the catalyst solution, then incubated with Ni-NTA agarose beads (from 150 µL of slurry) at 4 °C for 4 hours. The flow-through was collected and the column was washed (4 mL, PBS pH 8.0, 50 mM, 0.3 M NaCl, 12.5 mM imidazole) to remove the excess polymer. The product was eluted from the column (∼1 mL, PBS pH 8.0, 50 mM, 0.3 M NaCl, 250 mM imidazole), dialysed against PBS (pH 8.0, 50 mM, 0.1 M NaCl), and concentrated to ∼210 µL.
The obtained product was purified by FPLC on a Superdex 75 column in PBS (pH 8.0, 50 mM, 0.1 M NaCl). The fractions eluted between 1.05–1.20 mL were collected and concentrated to ∼110 µL.
DLS measurements
A solution of the biohybrid (100 µL in PBS pH 8.0, 50 mM, 0.1 M NaCl) was placed in a disposable low volume plastic cuvette, which was tightly sealed with a wad of Parafilm a few millimetres above the surface of the solution in order to minimise evaporation during the heating cycles.A temperature trend measurement was performed from 15 to 55 °C, taking a measurement at every 1 °C. The sample was then maintained at 55 °C and a series of measurements were taken before performing the inverse temperature trend measurement (55 to 15 °C, measurement at every 1 °C) and taking measurements at 15 °C. This cycle was repeated a total of three times. Finally, the sample was cooled to 5 or 10 °C and low temperature measurements were performed.
TEM measurements
A solution of the biohybrid (50 µL in PBS pH 8.0, 50 mM, 0.1 M NaCl) and an aliquot of deionised water (15 µL) were placed in a water bath. A TEM grid was placed on aluminium foil floating on the surface of the same bath, which was covered in foil to help to maintain a constant temperature. The system was heated to 55 °C at a rate of 1 °C min−1, then allowed to equilibrate for 10 minutes. An aliquot of the biohybrid solution (3 µL) was added to the water using a warmed pipette tip and left to equilibrate for a minute. A drop of this solution (6 µL) was deposited on the TEM grid using a warmed pipette tip. The bath was tightly covered in foil to minimise evaporation and the drop was left on the grid for 5 minutes. The excess liquid was then removed with a filter paper and the grid was left to air dry at room temperature. The dilution in water and the removal of the excess liquid helped us to ensure that minimal amounts of salt were present on the prepared TEM grids. The samples were stained by placing a drop of uranyl acetate solution on the grid (5 µL, 0.2% (w/v) in deionised water) for 15 seconds, then removing it with filter paper. The grid was left to air dry at room temperature. Grids were visualised by TEM both before and after staining.Acknowledgements
We gratefully acknowledge the Netherlands Organisation for Scientific Research (NWO, Rubicon grant for C.L., Veni grant for R.H., and Vidi grant for J.J.L.M.) and the National Sciences and Engineering Research Council of Canada (NSERC, Post-doctoral scholarship for C.L.). R.J.M.N. acknowledges the Royal Netherlands Academy of Arts and Science for financial support and J.J.L.M. acknowledges the European Science Foundation (ESF) for an EURYI Grant.Notes and references
- S. F. M. van Dongen, H. P. M. de Hoog, R. J. R. W. Peters, M. Nallani, R. J. M. Nolte and J. C. M. van Hest, Chem. Rev., 2009, 109, 6212 CrossRef CAS.
- A. Carlsen and S. Lecommandoux, Curr. Opin. Colloid Interface Sci., 2009, 14, 329 CrossRef CAS.
- A. Choucair, C. Lavigueur and A. Eisenberg, Langmuir, 2004, 20, 3894 CrossRef CAS.
- L. F. Zhang and A. Eisenberg, Science, 1995, 268, 1728 CrossRef CAS.
- A. Blanazs, S. P. Armes and A. J. Ryan, Macromol. Rapid Commun., 2009, 30, 267 CrossRef CAS.
- D. E. Discher, V. Ortiz, G. Srinivas, M. L. Klein, Y. Kim, D. A. Christian, S. S. Cai, P. Photos and F. Ahmed, Prog. Polym. Sci., 2007, 32, 838 CrossRef CAS.
- A. J. Dirks, J. J. L. M. Cornelissen, F. L. van Delft, J. C. M. van Hest, R. J. M. Nolte, A. E. Rowan and F. P. J. T. Rutjes, QSAR Comb. Sci., 2007, 26, 1200 Search PubMed.
- K. L. Heredia and H. D. Maynard, Org. Biomol. Chem., 2007, 5, 45 RSC.
- I. C. Reynhout, J. J. L. M. Cornelissen and R. J. M. Nolte, Acc. Chem. Res., 2009, 42, 681 CrossRef CAS.
- P. Thordarson, B. Le Droumaguet and K. Velonia, Appl. Microbiol. Biotechnol., 2006, 73, 243 CrossRef CAS.
- B. Le Droumaguet, G. Mantovani, D. M. Haddleton and K. Velonia, J. Mater. Chem., 2007, 17, 1916 RSC.
- M. J. Boerakker, N. E. Botterhuis, P. H. H. Bomans, P. M. Frederik, E. M. Meijer, R. J. M. Nolte and N. A. J. M. Sommerdijk, Chem.–Eur. J., 2006, 12, 6071 CrossRef CAS.
- M. J. Boerakker, J. M. Hannink, P. H. H. Bomans, P. M. Frederik, R. J. M. Nolte, E. M. Meijer and N. A. J. M. Sommerdijk, Angew. Chem., Int. Ed., 2002, 41, 4239 CrossRef CAS.
- A. J. T. Dirks, S. S. van Berkel, N. S. Hatzakis, J. A. Opsteen, F. L. van Delft, J. J. L. M. Cornelissen, A. E. Rowan, J. C. M. van Hest, F. P. J. T. Rutjes and R. J. M. Nolte, Chem. Commun., 2005, 4172 RSC.
- J. M. Hannink, J. J. L. M. Cornelissen, J. A. Farrera, P. Foubert, F. C. De Schryver, N. A. J. M. Sommerdijk and R. J. M. Nolte, Angew. Chem., Int. Ed., 2001, 40, 4732 CrossRef CAS.
- B. Le Droumaguet and K. Velonia, Angew. Chem., Int. Ed., 2008, 47, 6263 CrossRef CAS.
- I. C. Reynhout, J. J. L. M. Cornelissen and R. J. M. Nolte, J. Am. Chem. Soc., 2007, 129, 2327 CrossRef CAS.
- K. Velonia, A. E. Rowan and R. J. M. Nolte, J. Am. Chem. Soc., 2002, 124, 4224 CrossRef CAS.
- F. Liu and M. W. Urban, Prog. Polym. Sci., 2010, 35, 3 CrossRef CAS.
- A. Kumar, A. Srivastava, I. Y. Galaev and B. Mattiasson, Prog. Polym. Sci., 2007, 32, 1205 CrossRef CAS.
- R. Pelton, Adv. Colloid Interface Sci., 2000, 85, 1 CrossRef CAS.
- Y. Xia, N. A. D. Burke and H. D. H. Stover, Macromolecules, 2006, 39, 2275 CrossRef CAS.
- C. Wu, Polymer, 1998, 39, 4609 CrossRef CAS.
- S. Hirotsu, Y. Hirokawa and T. Tanaka, J. Chem. Phys., 1987, 87, 1392 CrossRef CAS.
- D. Neugebauer, Polym. Int., 2007, 56, 1469 CrossRef CAS.
- Z. B. Hu, T. Cai and C. L. Chi, Soft Matter, 2010, 6, 2115 RSC.
- J. F. Lutz, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3459 CrossRef CAS.
- J. F. Lutz, O. Akdemir and A. Hoth, J. Am. Chem. Soc., 2006, 128, 13046 CrossRef CAS.
- C. R. Becer, S. Hahn, M. W. M. Fijten, H. M. L. Thijs, R. Hoogenboom and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7138 CrossRef CAS.
- K. Skrabania, J. Kristen, A. Laschewsky, O. Akdemir, A. Hoth and J. F. Lutz, Langmuir, 2007, 23, 84 CrossRef CAS.
- D. Popescu, R. Hoogenboom, H. Keul and M. Moeller, Polym. Chem., 2010, 1, 878 RSC.
- W. Steinhauer, R. Hoogenboom, H. Keul and M. Moeller, Macromolecules, 2010, 43, 7041 CrossRef CAS.
- V. Coessens, T. Pintauer and K. Matyjaszewski, Prog. Polym. Sci., 2001, 26, 337 CrossRef CAS.
- A. S. Hoffman and P. S. Stayton, Macromol. Symp., 2004, 207, 139 CrossRef CAS.
- A. S. Hoffman and P. S. Stayton, Prog. Polym. Sci., 2007, 32, 922 CrossRef CAS.
- P. De, M. Li, S. R. Gondi and B. S. Sumerlin, J. Am. Chem. Soc., 2008, 130, 11288 CrossRef CAS.
- K. L. Heredia, D. Bontempo, T. Ly, J. T. Byers, S. Halstenberg and H. D. Maynard, J. Am. Chem. Soc., 2005, 127, 16955 CrossRef CAS.
- S. Kulkarni, C. Schilli, B. Grin, A. H. E. Muller, A. S. Hoffman and P. S. Stayton, Biomacromolecules, 2006, 7, 2736 CrossRef CAS.
- S. Kulkarni, C. Schilli, A. H. E. Muller, A. S. Hoffman and P. S. Stayton, Bioconjugate Chem., 2004, 15, 747 CrossRef CAS.
- M. Li, P. De, S. R. Gondi and B. S. Sumerlin, Macromol. Rapid Commun., 2008, 29, 1172 CrossRef CAS.
- M. Li, P. De, H. Li and B. S. Sumerlin, Polym. Chem., 2010, 1, 854 RSC.
- H. D. Maynard, K. L. Heredia, R. C. Li, D. P. Parra and V. Vazquez-Dorbatt, J. Mater. Chem., 2007, 17, 4015 RSC.
- S. Salmaso, S. Bersani, S. S. Pennadam, C. Alexander and P. Caliceti, Int. J. Pharm., 2007, 340, 20 CrossRef CAS.
- V. Vazquez-Dorbatt, Z. P. Tolstyka and H. D. Maynard, Macromolecules, 2009, 42, 7650 CrossRef CAS.
- A. J. Dirks, J. J. L. M. Cornelissen and R. J. M. Nolte, Bioconjugate Chem., 2009, 20, 1129 CrossRef CAS.
- K. Sivakumar, F. Xie, B. M. Cash, S. Long, H. N. Barnhill and Q. Wang, Org. Lett., 2004, 6, 4603 CrossRef.
- B. Karlen, B. Lindeke, S. Lindgren, K. G. Svensson, R. Dahlbom, D. J. Jenden and J. E. Giering, J. Med. Chem., 1970, 13, 651 CrossRef CAS.
- T. R. Chan, R. Hilgraf, K. B. Sharpless and V. V. Fokin, Org. Lett., 2004, 6, 2853 CrossRef CAS.
- S. I. Van Kasteren, H. B. Kramer, D. P. Gamblin and B. G. Davis, Nat. Protoc., 2007, 2, 3185 Search PubMed.
- I. J. Minten, L. J. A. Hendriks, R. J. M. Nolte and J. J. L. M. Cornelissen, J. Am. Chem. Soc., 2009, 131, 17771 CrossRef CAS.
Footnotes |
| † This paper is part of a Polymer Chemistry issue highlighting the work of emerging investigators in the polymer chemistry field. Guest Editors: Rachel O'Reilly and Andrew Dove. |
| ‡ Electronic supplementary information (ESI) available: Full characterisation of the polymers, turbidimetry studies with varying polymer or salt concentration and multiple cycles study, mass spectra characterisation of acetylene-functionalised EGFP, and FPLC traces of the biohybrids after purification by FPLC. See DOI: 10.1039/c0py00229a |
| This journal is © The Royal Society of Chemistry 2011 |