Design of AB divinyl “template monomers” toward alternating sequence control in metal-catalyzed living radical polymerization†‡
Yusuke
Hibi
,
Shinsuke
Tokuoka
,
Takaya
Terashima
,
Makoto
Ouchi
* and
Mitsuo
Sawamoto
*
Department of Polymer Chemistry, Graduate School of Engineering, Kyoto University, Katsura, Nishikyo-ku, Kyoto 615-8510, Japan. E-mail: ouchi@living.polym.kyoto-u.ac.jp; sawamoto@star.polym.kyoto-u.ac.jp; Fax: +81-75-383-2601; Tel: +81-75-383-2601
First published on 27th September 2010
Abstract
For alternating repeat-unit sequence via living radical polymerization, “template monomers” were designed and polymerized, where two polymerizable alkene (vinyl) functions [e.g., methacrylate (M) and acrylate (A)] were placed side by side at the 1,8-positions on a rigid naphthalene scaffold. Even for such a divinyl monomer, highly selective intramolecular radical propagation was achieved with metal-catalyzed living radical polymerization systems, to give linear controlled polymers without cross-linking. The naphthalene template was cleaved viahydrolysis from the resultant polymer, and subsequently methylated for sequence characterization. 1H NMR analysis demonstrated that the polymers consisted of highly alternating sequences (A-M-A: >80%), practically free from homo triad sequences (M-M-M).
Introduction
“Sequence” has recently drawn attention as a next-targeted structural factor to be precisely controlled in synthetic macromolecules.1–11 Obviously, their role models in nature include DNA or protein, whose sequence of constitutional repeat units is perfectly controlled and defined to form unique three-dimensional structures and, in turn, to express advanced functions. However, the sequence control in artificial polymer systems is still beyond our reach, and no efficient ways have been proposed or achieved.In radical polymerization, for example, copolymers consist of statistical repeat-unit sequences that depend on the reactive ratios (r) of monomers, as a function of the electronic and the steric characteristics of their double bonds. Mostly, there is no selectivity of growing radical species for next monomer to give random sequence, except for specific monomer combination giving “alternating copolymerization”, such as vinyl ether/malic anhydride and styrene/maleimide. It is considerably difficult to achieve “selectivity control” that ultimately leads to “sequence control”, although addition of a Lewis acid is known to change the copolymerizability of ester/amide-based monomers through modification of the electron density on their vinyl groups.12
On the other hand, we have achieved “reactivity control” via metal-mediated living radical polymerization in which side reactions (i.e., irreversible terminations and chain transfer reactions) are well suppressed.13–17 It is crucial to reversibly soothe “active” radical species to become “dormant” by using a capping agent and to trigger a dynamic equilibrium by such stimuli as heat, light, and catalyst. Now, we can control the chain length (molecular weight) and terminal structures of macromolecules and can thus construct well-defined polymeric architectures such as block, star, and graft copolymers. However, the copolymerizability in living polymerization is virtually the same as that in conventional radical copolymerization, since the essential properties of radical species are identical regardless of their livingness. Some additional strategy would be required to change or control copolymerizability for sequence control.
Based on this background, we designed template initiators for metal-catalyzed living radical polymerization toward sequence control (Scheme 1A)6 that would mimic sophisticated template systems in nature. In this strategy, template molecules carrying recognition tags are introduced via living cationic polymerization, into a position within the initiator close to the initiating site for metal-catalyzed living radical polymerization, so as to induce specific recognition of a particular monomer and thereby selective radical propagation of “recognized” monomers. Though still primitive, we have in fact achieved the selective radical addition reaction of methacrylic acid, a monomer with a pendent carboxylic acid to be specifically “recognized by the template amino group over non-interacting monomers (e.g., methyl methacrylate). This result tells us that a concept with template molecules is possibly an efficient approach towards sequence-controlled radical polymerization.
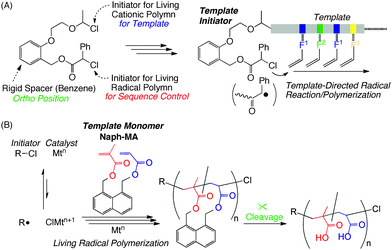 | ||
| Scheme 1 Template systems towards sequence controlled radical polymerization with a template initiator (A) and a template monomer (B: this work) | ||
In this work, our effort to develop the template concept is now directed at the design of monomers: two kinds of vinyl monomers, i.e., methacrylate (M) and acrylate (A), are immobilized on the peri-position of naphthalene in advance as a rigid scaffold template [hereafter “divinyl template monomer”; Naph-MA in Scheme 1B]. In our strategy, the designer divinyl template monomer is polymerized into soluble linear polymers by metal-catalyzed living radical polymerization (i.e., cyclopolymerization18), under diluted conditions to avoid undesired cross-linking reactions. The ester linkages (spacers) in the resultant polymer are subsequently cleaved viahydrolysis to give copolymer of corresponding (meth)acrylic acids. A repetitive sequential propagation on the naphthalene template would enhance alternating sequence of the methacrylate and the acrylate monomers. To our knowledge, this is a first attempt to apply a cyclopolymerization for sequence control.
A platform system for controlled polymerization was first studied using a methacrylate-based homo divinyl template monomer (Naph-MM) to achieve living radical (cyclo)polymerization without cross-linking reactions. These conditions were then applied to the polymerization of Naph-MA, and the naphthalene templates were cleaved from the resultant polymer to characterize the sequence.
Herein we report that, even for these divinyl monomers, controlled polymerizations were achieved with a suitable ruthenium catalyst under diluted conditions ([monomer]0 < 100 mM) to give well-defined linear polymers. Structural analysis by 1H NMR showed that the copolymers from Naph-MA was clearly rich in alternating triad sequence (> 80%) and clearly differs in sequence from conventional random copolymers of methyl methacrylate and methyl acrylate.
Experimental
Materials
Measurements
The molecular weight distribution, Mn, and Mw/Mn values of polymers measured in chloroform at 40 °C on three polystyrene gel columns [Shodex K-805L (pore size: 20–1000 Å; 8.0 mm i.d. × 30 cm) × 3; flow rate 1.0 mL min−1] connected to a Jasco PU-980 precision pump and a Jasco 930-RI refractive-index detector, and 970-UV ultraviolet detector. The columns were calibrated against 13 standard PSt samples (Polymer Laboratories; Mn = 630–1,200,000, Mw/Mn = 1.06–1.22) as well as the monomer. 1H-NMR spectra of the obtained polymers were recorded in CDCl3 at 25 °C on a JEOL JNM-LA500 spectrometer operating at 500.16 MHz. Polymers for 1H-NMR analysis were fractionated by preparative SEC (column: Shodex K-2002). MALDI-TOF-MS analysis was performed on a Shimadzu AXIMA-CFR instrument equipped with 1.2 m linear flight tubes and a 337 nm nitrogen laser; the matrix was dithranol.Monomer syntheses
 | ||
| Scheme 2 Syntheses of template monomers (Naph-MM and Naph-MA) | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, followed by toluene–AcOEt = 9:1 to afford Naph-M as an oil in 60% yield (3.2 g). To a solution of Naph-M (3.2 g, 13 mmol) in dichloromethane (50 mL) were added triethylamine (4.4 mL, 32 mmol) and acryloylchloride (2.6 mL, 32 mmol) at 0 °C. The resulting mixture was stirred at room temperature for 4 h. The reaction was quenched by adding H2O and the mixture was partitioned between EtOAc and H2O. The aqueous layer was extracted 3 times with EtOAc. The combined EtOAc solution was washed with brine, dried on Na2SO4 and evaporated. The residue was chromatographed on silica gel eluting hexane–AcOEt = 9:1 to afford Naph-MM as an oil in 80% yield (3.1 g).
:1, followed by toluene–AcOEt = 9:1 to afford Naph-M as an oil in 60% yield (3.2 g). To a solution of Naph-M (3.2 g, 13 mmol) in dichloromethane (50 mL) were added triethylamine (4.4 mL, 32 mmol) and acryloylchloride (2.6 mL, 32 mmol) at 0 °C. The resulting mixture was stirred at room temperature for 4 h. The reaction was quenched by adding H2O and the mixture was partitioned between EtOAc and H2O. The aqueous layer was extracted 3 times with EtOAc. The combined EtOAc solution was washed with brine, dried on Na2SO4 and evaporated. The residue was chromatographed on silica gel eluting hexane–AcOEt = 9:1 to afford Naph-MM as an oil in 80% yield (3.1 g).
Polymerization
Sequence analysis
Results and discussion
Controlled radical polymerization of Naph-MM
For cyclopolymerization of divinyl template monomers, it is essential to apply a low monomer concentration, to control sequential and intramolecular dimeric propagation on the naphthalene template over intermolecular growth (cross-linking). Thus, we first studied the effects of monomer concentration on polymerization behaviors using the homo divinyl template monomer, Naph-MM.Polymerization of Naph-MM was performed with H–(MMA)2–Cl (initiator), Cp*RuCl(PPh3)2 (catalyst), and n-Bu2NH (cocatalyst) in toluene at 80 °C under the so-called 10-mer conditions ([Naph-MM]0/[initiator]0 = 10; 20 equivalents for the vinyl group). The polymerization at [Naph-MM]0 = 250 mM resulted in gelation within 20 min but remained homogeneous throughout at lower concentrations ([Naph-MM]0 = 100 and 50 mM) and the methacrylate’s vinyl group was smoothly consumed (by 1H NMR; Fig. 1). SEC curves of the produced polymers were narrow and unimodal (Mw/Mn = 1.15–1.35) and shifted to higher molecular weight as the reaction proceeded. Thus, it was possible to control radical propagation even with the divinyl monomer immobilized on a naphthalene template without cross-linking by lowering the monomer concentration.
![The effects of monomer concentration on ruthenium-catalyzed living radical polymerization of Naph-MM in toluene at 60 °C: [Naph-MM]0/[H–(MMA)2–Cl]0/[Cp*RuCl(PPh3)2]0/n-Bu2NH = 250/25/2.5/100, 100/10/1.0/40, or 50/5.0/0.5/20 mM.](/image/article/2011/PY/c0py00252f/c0py00252f-f1.gif) | ||
| Fig. 1 The effects of monomer concentration on ruthenium-catalyzed living radical polymerization of Naph-MM in toluene at 60 °C: [Naph-MM]0/[H–(MMA)2–Cl]0/[Cp*RuCl(PPh3)2]0/n-Bu2NH = 250/25/2.5/100, 100/10/1.0/40, or 50/5.0/0.5/20 mM. | ||
To increase the degree of polymerization of products, the 25-mer condition ([Naph-MM]0/[initiator]0 = 100/4 mM; 50 equivalent for vinyl group) was next targeted, and solvent effects were also examined (Fig. 2). In toluene, the monomer was more rapidly consumed (conv.= 93% in 12 h), but the system turned into heterogeneous at conversion ∼ 60%. The worse control would have been due to the higher local concentration of Naph-MM and the growing poly(Naph-MM) chains that are poorly soluble in toluene. In contrast, polymerization systems appeared homogeneous in anisole and 1,2-dichloroethane (DCE), and the conversions reached over 90%. Molecular weights of the produced polymers were fairly controlled with narrow molecular weight distributions (Mw/Mn = 1.15–1.30).
![Effects of solvent on ruthenium-catalyzed living radical polymerization of Naph-MM at 60 °C: [Naph-MM]0/[H–(MMA)2–Cl]0/[Cp*RuCl(PPh3)2]0/n-Bu2NH = 100/4/0.4/16 mM.](/image/article/2011/PY/c0py00252f/c0py00252f-f2.gif) | ||
| Fig. 2 Effects of solvent on ruthenium-catalyzed living radical polymerization of Naph-MM at 60 °C: [Naph-MM]0/[H–(MMA)2–Cl]0/[Cp*RuCl(PPh3)2]0/n-Bu2NH = 100/4/0.4/16 mM. | ||
The structures of poly(Naph-MM)s were analyzed in detail by MALDI-TOF-MS for samples obtained in anisole and DCE [Mn (Mw/Mn) = 2500 (1.16) and 4000 (1.18), respectively] (Fig. 3). In both cases, a common series was observed, which consisted of regularly spaced peaks with the Naph-MM mass interval (324.4), and each peak agreed with that for the anticipated linear living polymer [H–(MMA)2–(Naph-MM)n–Cl + Na+]. However, the sample from anisole also exhibited another minor series with a higher peak mass shifted from the major series by the mass of the initiator [H–(MMA)2–Cl: MW = 236.7], indicating branches carrying two initiator moieties. The difference in polymer structure would be related to different solubility of the monomer and the polymers in the two solvents, and DCE appears a better solvent for Naph-MM and poly(Naph-MM) than toluene and anisole. Thus, the higher solubility (or homogeneity) would be important to suppress cross-linking.
 | ||
| Fig. 3 MALDI-TOF-MS spectra of poly(NaPh-MM) obtained in anisole (A) and in DCE. See Fig. 2 caption for the polymerization condition. | ||
Tacticity of poly(Naph-MM)
The polymerization of Naph-MM is regarded as a kind of cyclopolymerization, where the strained conformation of the growing species on the template (spacer) framework is known to induce unusual stereospecificity.23,24 To examine such template effects on stereospecificity, the tacticity of poly(Naph-MM) was analyzed. A completely soluble sample obtained in DCE (Mn = 5900; Mw/Mn = 1.19) was hydrolyzed into poly(methaclylic acid) (see ESI†) and the resulting acid polymer was subsequently methylated with trimethylsilyl-diazomethane (TMSCH2N2) into poly(methyl methacylate) (PMMA). 1H NMR analysis for α- methyl proton (split peaks at 0.8–1.2 ppm) indicated a heterotactic-rich tacticity (mm/mr/rr = 2/63/35%: see ESI†). For comparison, benzyl methacrylate, a non-template model monomer, was also polymerized in DCE under otherwise the same conditions, and the polymer (Mn = 3400 and Mw/Mn = 1.36) was similarly converted into PMMA. The tacticity was rather syndiotactic-rich (mm/mr/rr = 5/34/61%), similar to conventional methacrylate-based polymersviaradical polymerization. The unusual tacticity would also be derived from the naphthalene template scaffold.Controlled radical polymerization of Naph-MA
With these initial results, controlled polymerization of the hetero divinyl template monomer (Naph-MA) was examined with ruthenium-catalyzed systems under similarly diluted conditions. Since the use of n-Bu2NH (cocatalyst) was found to cause undesired reactions for the acrylate terminal, a ruthenium catalyst without cocatalyst was employed: a bisphosphine monoxide-ligated Cp*-ruthenium with 1,2-bis(diphenylphosphino)ethane monoxide (PO–2) as ligand (Cp*: pentamethylcyclopentadiene).20 The polymerization condition is as follows: [Naph-MA]0/[H–(MMA)2–Cl]0/[[Cp*Ru(μ3-Cl)]4]0/[PO–2]0 = 50/2.0/0.125/1.0 mM, in DCE at 80 °C.For Naph-MA the methacrylate and the acrylate vinyl groups could separately be monitored by 1H NMR. Both were smoothly consumed in parallel with each other, and finally the conversions reached over 80% in 160 h (Fig. 4A). The system was homogeneous throughout, and the obtained polymers were fairly controlled in molecular weight (Mw/Mn ∼ 1.40: Fig. 4B). The MALDI-TOF-MS spectrum of the polymer (Mn = 2,900, Mw/Mn = 1.45) showed a single series of regularly spaced peaks with an interval of the Naph-MA mass (MW = 310.3) (Fig. 4C). Each peak mass was in good agreement with that for the expected living polymer [H–(MMA)2–(Naph-MA)n–Cl + Na+]. Thus, controlled radical cyclopolymerization of Naph-MA was achieved with the ruthenium catalyst system.
![Ruthenium-catalyzed living radical polymerization of Naph-MA in DCE at 80 °C: [Naph-MA]0/[H–(MMA)2–Cl]0/[[Cp*Ru(μ3-Cl)]4]0/[1,2-bis(diphenylphosphino)ethane monoxide (PO–2)] = 50/2.0/0.125/1.0 mM. (A) Time-conversion, (B) SEC curves, (C) MALDI-TOF-MS spectrum.](/image/article/2011/PY/c0py00252f/c0py00252f-f4.gif) | ||
| Fig. 4 Ruthenium-catalyzed living radical polymerization of Naph-MA in DCE at 80 °C: [Naph-MA]0/[H–(MMA)2–Cl]0/[[Cp*Ru(μ3-Cl)]4]0/[1,2-bis(diphenylphosphino)ethane monoxide (PO–2)] = 50/2.0/0.125/1.0 mM. (A) Time-conversion, (B) SEC curves, (C) MALDI-TOF-MS spectrum. | ||
Template cleavage and sequence analysis
Finally, the repeat-unit sequence of poly(Naph-MA) (Mn = 4,300, Mw/Mn = 1.37: see Fig. 4B) was analyzed by 1H NMR. The naphthalene templates along the backbone was first cleaved by hydrolysis with CF3COOH, and the resultant acid polymer was methylated with TMSCH2N2 into a copolymer of methyl methacrylate and methyl acrylate (Scheme 3). As shown in Fig. 5, the multiple peaks at 0.8–1.2 ppm, assigned to the MMA α-methyl protons, provided information about the triad sequence and tacticity for MMA-centered units (X-M-X).25 These peaks and their relative intensities were compared with those for the corresponding statistical random copolymer of MMA and MA.26 | ||
| Scheme 3 Template cleavage and subsequent methylation for poly(Naph-MA) | ||
 | ||
| Fig. 5 Sequence analyses by 1H NMR spectra (CDCl3, r.t.) with α-methyl proton of methyl methacrylate unit at 0.80–1.24 ppm. (A) copolymervia 1:1 radical copolymerization of methyl methacrylate and methylacrylate (B) copolymer after hydrolysis and methylation of poly(Naph-MA). | ||
The most striking difference between the two samples was seen in a peak at higher field (6 at ∼0.85 ppm), assigned to syndiotactic M-M-M sequence [M-M-M(S)]: 21% (random) vs. ∼0% (Naph-MA). The absence of the M-M-M triad MMA homo-sequence for the poly(Naph-MA) makes sense of the controlled polymerization without cross-linking (see above). It is particularly worth noting that, in this cyclopolymer sample relative to the random copolymer, peak 5 (∼ 0.91 ppm), assigned to M-M-A and A-M-M sequences (both heterotactic and syndiotactic), was much weaker (∼10%), while the peaks 2, 3, and 4 for the alternatingA-M-A sequence were obviously stronger. The alternating sequence dominated roughly over 80% of the total enchainment, which means that at least 18 out of 20 propagation steps were regulated to the MMA → MA intramolecular growth direction, as the template dictates; under the 25-mer conditions, the observed conversion of 80% implies propagation has been repeated 20 times on average.
The observed high selectivity for both intramolecular cyclopropagation and alternating sequence indicates that the methacrylate group, more reactive than acrylate, would preferentially or selectively react relative to the adjacent acrylate group on the same template. Given a small difference in reactivity between the two vinyl units, such a high selectivity would stem from the fact that, in the Naph-MA system, the concentrations of methacrylate and acrylate are by definition kept equal in a dilute solution during polymerization. This situation totally differs from that in conventional random copolymerization, where the instantaneous local concentrations of comonomers (fed at 1:1 ratio) vary even though the apparent consumption rate is same and the initial feed ratio. These results suggest that design of template monomer in consideration of reactivity would open a door to more sophisticated sequence control.
Conclusions
Advanced sequence control for alternating repeat-unit arrays was realized in radical polymerization with a designer “template monomer”: two types of vinyl monomer units, methacrylate and acrylate, were embedded at the peri-position of a naphthalene template. One crucial factor for the success of this concept is, as it turned out in this work, a reaction control for the cyclopolymerization on the naphthalene without cross-linking, which was achieved with metal-catalyzed living radical polymerization under relatively diluted conditions. Another factor is a reactivity difference between the embedded two polymerizable units, and, interestingly, the template framework allowed high alternating sequence via repetitive regular propagation for the methacrylate-acrylate pair whose reactivity difference per se would be too small to induce such an alternating sequence.Acknowledgements
This research was partially supported by the Ministry of Education, Science, Sports and Culture through a Grant-in-Aid for Creative Science Research (18GS0209) for which the authors are grateful.Notes and references
- N. Badi and J. F. Lutz, Chem. Soc. Rev., 2009, 38, 3383–3390 RSC.
- J. F. Lutz, Polym. Chem., 2010, 1, 55–62 RSC.
- J. F. Lutz, Nat. Chem., 2010, 2, 84–85 Search PubMed.
- S. Pfeifer and J. F. Lutz, J. Am. Chem. Soc., 2007, 129, 9542–9543 CrossRef CAS.
- S. Pfeifer and J. F. Lutz, Chem.–Eur. J., 2008, 14, 10949–10957 CrossRef CAS.
- S. Ida, T. Terashima, M. Ouchi and M. Sawamoto, J. Am. Chem. Soc., 2009, 131, 10808–10809 CrossRef CAS.
- R. E. Kleiner, Y. Brudno, M. E. Birnbaum and D. R. Liu, J. Am. Chem. Soc., 2008, 130, 4646–4659 CrossRef CAS.
- J. W. Kramer, D. S. Treitler, E. W. Dunn, P. M. Castro, T. Roisnel, C. M. Thomas and G. W. Coates, J. Am. Chem. Soc., 2009, 131, 16042–16043 CrossRef CAS.
- K. Satoh, S. Ozawa, M. Mizutani, K. Nagai and M. Kamigaito, Nat. Commun., 2010, 1, 1–6 Search PubMed.
- K. Satoh, M. Matsuda, K. Nagai and M. Kamigaito, J. Am. Chem. Soc., 2010, 132, 10003–10005 CrossRef CAS.
- R. M. Stayshich and T. Y. Meyer, J. Am. Chem. Soc., 2010, 132, 10920–10934 CrossRef CAS.
- M. Hirooka, H. Yabuuchi, J. Iseki and Y. Nakai, J. Polym. Sci., Part A-1, 1968, 6, 1381–1396 CrossRef CAS.
- M. Kamigaito, T. Ando and M. Sawamoto, Chem. Rev., 2001, 101, 3689–3745 CrossRef CAS.
- M. Kamigaito, T. Ando and M. Sawamoto, Chem. Rec., 2004, 4, 159–175 CrossRef CAS.
- M. Ouchi, T. Terashima and M. Sawamoto, Acc. Chem. Res., 2008, 41, 1120–1132 CrossRef CAS.
- M. Ouchi, T. Terashima and M. Sawamoto, Chem. Rev., 2009, 109, 4963–5050 CrossRef CAS.
- K. Matyjaszewski and J. H. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS.
- G. B. Butler, Acc. Chem. Res., 1982, 15, 370–378 CrossRef CAS.
- T. Ando, M. Kamigaito and M. Sawamoto, Macromolecules, 1998, 31, 6708–6711 CrossRef CAS.
- Y. Fukuzaki, Y. Tomita, T. Terashima, M. Ouchi and M. Sawamoto, Macromolecules, 2010, 43, 5989–5995 CrossRef CAS.
- H. Luo, Q. Zeng, Z. R. Liu, Y. Z. Wei, B. G. Li and F. P. Wang, Synth. Commun., 2004, 34, 2269–2275 CrossRef CAS.
- K. Ishitake, K. Satoh, M. Kamigaito and Y. Okamoto, Angew. Chem., Int. Ed., 2009, 48, 1991–1994 CrossRef CAS.
- T. Kakuchi, H. Kawai, S. Katoh, O. Haba and K. Yokota, Macromolecules, 1992, 25, 5545–5546 CrossRef CAS.
- R. Saito and K. Yamaguchi, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 6262–6271 CrossRef CAS.
- M. M. C. Lopezgonzalez, M. Fernandezgarcia, J. M. Barralesrienda, E. L. Madruga and C. Arias, Polymer, 1993, 34, 3123–3128 CrossRef CAS.
- The copolymer was obtained with ruthenium-catalyzed living radical copolymerization of MMA and MA in toluene at 80 °C: [MMA]0/[MA]0/[H–(MMA)2–Cl]0/[[Cp*Ru(μ3-Cl)]4]0/[PO-2]0 = 2000/2000/40/1.0/8.0 mM. Conversion: 100% (MMA); 90% (MA). Mn = 10,800, Mw/ Mn = 1.19.
Footnotes |
| † This paper is part of a Polymer Chemistry issue highlighting the work of emerging investigators in the polymer chemistry field. Guest Editors: Rachel O'Reilly and Andrew Dove. |
| ‡ Electronic supplementary information (ESI) available: Tacticity analysis with 1H NMR for poly(Naph-MM). See DOI: 10.1039/c0py00252f |
| This journal is © The Royal Society of Chemistry 2011 |