Polymeric cyclodextrin-based nanoparticles: synthesis, characterization and sorption properties of three selected pharmaceutically active ingredients
Pu
Xiao
a,
Yves
Dudal
b,
Philippe F.-X.
Corvini
a and
Patrick
Shahgaldian
*a
aSchool of Life Sciences, University of Applied Sciences Northwestern Switzerland, Gründenstrasse 40, 4132, Muttenz, Switzerland. E-mail: patrick.shahgaldian@fhnw.ch
bEnvolure, 2 place Pierre Viala, 34060, Montpellier Cedex 2, France
First published on 29th October 2010
Abstract
The reaction of three cyclodextrins, namely α-, β- and γ-cyclodextrins, with toluene-2,4-diisocyanate yields water-insoluble polymers that in the case of the α- and β-derivatives self-assemble in aqueous and ethanolic solutions as polydisperse polymeric nanoparticles. The so-produced nanomaterials have been characterized by means of scanning electron microscopy, BET (Brunauer, Emmett and Teller) nitrogen adsorption technique and ζ-potential measurements. The interactions of these nanoparticles with three selected pharmaceutically active ingredients (levofloxacin, aspirin and acetaminophen) have been studied in aqueous solution. It is demonstrated that in all cases the interactions of the studied drugs with the produced polymeric nanomaterials follow the well-established Freundlich model suggesting that the polymers act as heterogeneous sorbents in aqueous phase. It is demonstrated that the β-cyclodextrin-based polymer exhibits the highest sorption capacity for all three pharmaceuticals compared to the α- and γ-analogues, with a higher affinity for aspirin. The influence of the chemical structure of the guest molecule on the interactions of the produced nanoparticulate polymers is discussed.
Introduction
The design of polymeric nanomaterials possessing specific molecular recognition properties is a challenge of interest for various industrial fields ranging from biomedical and pharmaceutical applications to separation techniques and environmental applications, to name but a few.1 A number of synthetic approaches have been developed to create materials possessing specific binding ability for defined target molecules. A possible approach to produce this kind of materials makes use of the molecular imprinting principle. It is based on the complex formation of monomers with a template molecule prior to polymerization, enabling the three dimensional positioning of the recognition units in the produced polymer.2,3 In addition to these template-assisted produced polymers, another possible approach for the design of materials possessing specific molecular recognition ability consists in introducing in the structure of the polymer, receptors with the desired recognition properties. Since the early 1990s and the development of supramolecular chemistry,4 a wide range of synthetic receptors have been designed and shown to possess recognition properties for guests ranging from small ionic entities to large biomolecules. Among these macrocycles, cyclodextrins (CDs) have attracted a special interest because of their favourable chemical structure.5,6CDs are cyclic oligosaccharides consisting of 6, 7 or 8 D-glucopyranose units, for α-, β- and γ-CD, respectively.6,7 With a hydrophobic cavity and two hydrophilic rims, CDs have a remarkable capacity to form inclusion complexes with a wide range of molecular targets.8,9 In addition, the 3D structure of these macrocycles, exhibiting all the primary alcohol functions on one rim and the secondary alcohols on the opposite rim, makes their regio-selective modification possible. As a matter of fact, cyclodextrin chemistry is since more than three decades a fertile playground for synthetic chemists and a wide variety of chemically modified cyclodextrins have been produced.6,7 For their application as heterogeneous sorbents in water, they are usually modified to prepare water insoluble systems; for instance, they can be immobilized on carrier materials. The modification of nanoparticles with CDs have been extensively studied by Kaifer et al. who demonstrated, for instance, that CD-capped Pd nanoparticles are effective catalysts for the hydrogenation of water-soluble alkenes.10 Another possible approach is the production of polymers using CDs as monomers. Crosslinkers such as epichlorohydrin,11hexamethylene diisocyanate12 or 4,4′-methylenebis-phenyldiisocyanate9 have been successfully used to prepare β-CD-based polymers, and these systems have been applied for the removal of dyes in water. Glutaraldehyde has also been used as a crosslinker to attach β-CD to polymers containing hydroxyl groups, and the adsorption capacities of the β-CD modified polymer for phenolic compounds have been demonstrated to be improved compared to that of the original polymer.13β-CD has also been crosslinked by different diisocyanates, and the obtained polymers have been shown to exhibit high sorption capacities for phenolic compounds.14–17 In addition, CD-based polymers have been used to complex phthalic acid esters,18aromatic compounds,8,19 and bicyclic organic compounds.20 Although many reports concern CD-based polymers as sorbents, there have been few on the production of these polymers in the form of nanoparticulate systems.21,22 One can expect that the improved surface-to-volume ratio confers a crucial advantage to these nanomaterials over the corresponding bulk polymers. These materials could meet a broad scope of applications such as the detection/removal of pharmaceuticals from water. The occurrence of pharmaceuticals in aquatic environments is currently receiving an increasing attention because of their potential adverse effects not only on ecosystems23 but also on human health.24,25 Solutions for their detection at trace concentration in environmental matrices and their efficient removal in wastewater treatment plants are needed. In the present manuscript, we report on the synthesis of a series of nanoparticulate CD-based polymers and their ability to bind three selected pharmaceuticals, namely levofloxacin (LVF), aspirin (ASP) and acetaminophen (APAP).
Experimental
Materials
α-CD and γ-CD have been purchased from CycloLab R&D Ltd. All other chemicals and solvents have been purchased from Sigma-Aldrich and used without further purification.Preparation of cyclodextrin-based polymers (CDPs)
CDPs (α-CDP, β-CDP and γ-CDP) have been prepared following a slightly modified method from that previously published by Hishiya et al.26 Briefly, CDs (α-CD, β-CD or γ-CD, 2.0 mmol) were dissolved in DMSO (60 mL), and toluene-2,4-diisocyanate (TDI) (1.22 g, 7.0 mmol) was added. The mixture was maintained under inert atmosphere and stirred at 65 °C for 3 h, poured into acetone to yield a white precipitate. The so-produced solids, CDPs, were filtered out and washed extensively with acetone (10 × 100 mL), hot water (10 × 100 mL) and hot ethanol (16 × 100 mL) to remove unreacted TDI and CD. Finally, the produced CDPs were dried under vacuum at 90 °C for 24 h prior to use.Characterization of CDPs
The morphologies of the CDPs were studied by scanning electron microscopy (SEM) using a Zeiss Supra 40VP system. 1 µL of a CDP suspension in ethanol was submitted to ultrasonic treatment (30 min, 35 kHz) and spread on a freshly cleaved mica slide. After evaporation of the solvent, the samples were (Au/Pd) sputter-coated for 15 s at 15 mA. The SEM micrographs were acquired with a InLens detector, using an accelerating voltage of 20 kV.ζ-Potentials were measured by electrophoretic mobility measurements using a Zetasizer Nano ZS system (Malvern Instruments Ltd.) and disposable capillary cells DTS 1060 (Malvern Instruments Ltd.). The samples were prepared by adding 5 mg CDPs to 6 mL sodium phosphate buffer (1 mM, pH = 7). The measurements were done in triplicates to ensure the reproducibility of the results.
The surface area of the produced CDPs was measured by the Brunauer–Emmett–Teller (BET) method using a Gemini V Analyser (Micromeritics Instrument Corp.).
Interaction studies
The sorption of three pharmaceuticals (LVF, ASP or APAP) to the produced CDPs was investigated in aqueous solution using increasing amounts of pharmaceuticals. 20 mg of CDPs were added to the 2 mL pharmaceutical aqueous solutions (4.0 × 10−5 to 3.0 × 10−3 mol L−1). The mixture was shaked at 20 °C for 16 h and the CDPs/pharmaceutical complexes were then separated by centrifugation at 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 10 min. 20 µL of the supernatant were then collected and injected in a high performance liquid chromatography system (HPLC, Agilent 1100 Series) equipped with a column (150 × 4.6 mm) packed with 3.0 µm PRONTOSIL 120-3-C18-SH (Bischoff-Chromatography, Germany). The flow rate of the mobile phase was 1.0 mL min−1. The detection of the isocratically eluted compounds was operated at 330, 226 and 246 nm for LVF (acetonitrile/water, 30:70), ASP (acetonitrile/water, 30:70) and APAP (acetonitrile/water, 20:80), respectively. The adsorption kinetics experiments were carried out suspending 20 mg of the CDPs in 2 mL of water. The appropriate amount of the pharmaceuticals were added and the mixture incubated at 20 °C under shaking. After 5, 10, 20, 40, 60, 120, 180 and 240 min, 10 µL of the supernatant were sampled from the mixture and the concentration of the pharmaceutical remaining free in solution was measured by HPLC. The pharmaceutical sorption capacity was defined as the percentage of pharmaceuticals adsorbed by the CDPs with respect to the starting amount of pharmaceuticals.
000g for 10 min. 20 µL of the supernatant were then collected and injected in a high performance liquid chromatography system (HPLC, Agilent 1100 Series) equipped with a column (150 × 4.6 mm) packed with 3.0 µm PRONTOSIL 120-3-C18-SH (Bischoff-Chromatography, Germany). The flow rate of the mobile phase was 1.0 mL min−1. The detection of the isocratically eluted compounds was operated at 330, 226 and 246 nm for LVF (acetonitrile/water, 30:70), ASP (acetonitrile/water, 30:70) and APAP (acetonitrile/water, 20:80), respectively. The adsorption kinetics experiments were carried out suspending 20 mg of the CDPs in 2 mL of water. The appropriate amount of the pharmaceuticals were added and the mixture incubated at 20 °C under shaking. After 5, 10, 20, 40, 60, 120, 180 and 240 min, 10 µL of the supernatant were sampled from the mixture and the concentration of the pharmaceutical remaining free in solution was measured by HPLC. The pharmaceutical sorption capacity was defined as the percentage of pharmaceuticals adsorbed by the CDPs with respect to the starting amount of pharmaceuticals.
Results and discussion
Three cyclodextrin-based polymers (CDPs) have been produced following a procedure slightly modified from that published by Hishiya et al.26 Briefly, CD (α, β or γ) is mixed in DMSO with a di-functional cross-linker, toluene-2,4-diisocyanate (TDI), cf.Fig. 1. After 3 hours of reaction, the mixture is poured onto acetone and thoroughly washed with acetone, water and ethanol. The produced polymers, CDPs, are urethane cross-linked cyclodextrins produced by the reaction of the isocyanate groups of TDI and the hydroxyl groups of the cyclodextrin. The isocyanate groups of TDI are known to react preferentially with the primary hydroxyl groups of the CDs. These macrocycles are therefore expected to be mainly unmodified at the secondary alcohol groups borne by the (wide) rims and therefore to have the ability to include organic guests inside their cavity.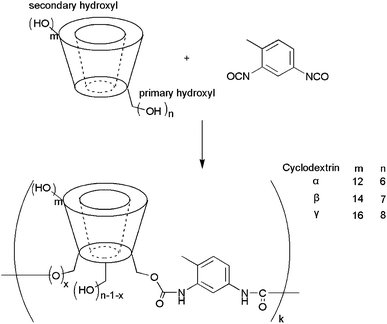 | ||
| Fig. 1 Preparation of cyclodextrin-based polymers. | ||
The morphology of the produced polymers have been studied using scanning electron microscopy; three characteristic micrographs of the produced CDPs, spread on a mica surface, are given in Fig. 2.
 | ||
| Fig. 2 SEM images of (a) α-CDP; (b) β-CDP; (c) γ-CDP. Scale bar: 100 nm. | ||
From these images, it could be clearly seen that the formed structures are aggregated nanoparticulate solids. Nevertheless, some major differences can be seen in terms of morphology. Indeed, while α-CDPs have quite a homogenous size distribution with diameters ranging from 50 to 120 nm, the NPs produced with β-CDP are more irregular in shape. γ-CDP is composed mainly of markedly irregular bigger aggregates. The differences observed might be attributed not only to the number of available hydroxyl groups for each cyclodextrin but also to reactivity or solubility differences. The ζ-potentials of α, β and γ-CDP were measured at −31.1 mV, −17.7 mV and −6.1 mV, respectively. The produced cyclodextrin polymer assemblies were also studied by photon correlation spectroscopy (PCS) experiments. The high polydispersity of the assemblies and their propensity to aggregate prevented us from obtaining reliable data. The BET surface areas of α, β and γ-CDP are 5.54 m2 g−1, 16.83 m2 g−1 and 7.52 m2 g−1, respectively. They increased in the order of α-CDP < γ-CDP < β-CDP.
Sorption isotherms
The binding capabilities of the produced CDPs with three selected pharmaceuticals (Fig. 3) have been studied by measuring the amount of remaining pharmaceuticals after incubation with the CDPs.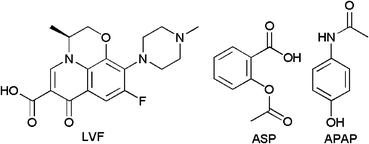 | ||
| Fig. 3 Structures of studied pharmaceuticals. | ||
In Fig. 4 are given the plots of the amount of bound pharmaceutical (Cs, mol g−1) per unit mass of CDPs versus the initial concentration of the tested pharmaceutical (Ci, mol L−1).
![Sorption isotherms of (a) LVF; (b) ASP and (c) APAP onto CDPs in solutions [CDP: 10 mg mL−1; contact time: 16 h].](/image/article/2011/PY/c0py00225a/c0py00225a-f4.gif) | ||
| Fig. 4 Sorption isotherms of (a) LVF; (b) ASP and (c) APAP onto CDPs in solutions [CDP: 10 mg mL−1; contact time: 16 h]. | ||
From Fig. 4, it could be seen that as expected the amount of the adsorbed pharmaceuticals increased with increasing the initial concentration of pharmaceutical. More interestingly, it can be seen that the binding capability of the β-CDP is significantly higher for all the tested APIs.
The sorption capacities of sorbents have been studied by applying different adsorption models (Langmuir, Freundlich, Langmuir–Freundlich, and Redlich–Petersen) and the experimental results have been shown to better fit to the Freundlich model suggesting the heterogeneous nature of the produced sorbents;27–29 the isotherms are given in Fig. 5.
![Freundlich sorption isotherms of (a) LVF; (b) ASP and (c) APAP onto CDPs in solutions [CDP: 10 mg mL−1; contact time: 16 h].](/image/article/2011/PY/c0py00225a/c0py00225a-f5.gif) | ||
| Fig. 5 Freundlich sorption isotherms of (a) LVF; (b) ASP and (c) APAP onto CDPs in solutions [CDP: 10 mg mL−1; contact time: 16 h]. | ||
The Freundlich model gives an empirical expression in the following form:
| Cs = KfCl1/n | (1) |
| CDP | LVF | ASP | APAP | ||||||
|---|---|---|---|---|---|---|---|---|---|
| K f | 1/n | r | K f | 1/n | r | K f | 1/n | r | |
| α | 0.008 | 0.774 | 0.999 | 0.065 | 1.092 | 0.981 | 0.009 | 0.910 | 0.999 |
| β | 0.024 | 0.840 | 0.999 | 0.587 | 1.218 | 0.982 | 0.023 | 0.881 | 0.999 |
| γ | 0.011 | 0.798 | 0.999 | 0.202 | 1.201 | 0.987 | 0.007 | 0.889 | 0.997 |
The higher value of Kf of β-CDP shows a better sorption capacity compared to that of α-CDP and γ-CDP. The results for APAP show the same trend that the data reported by Uekama et al. where the binding capacities of phenytoin to α, β or γ-CD–epichlorohydrin polymers (α, β or γ-CD·EP) increased such as: γ-CD·EP ≤ α-CD·EP < β-CD·EP.30 These results were attributed to the cavity size of β-CD better suited to complex phenytoin. Yilmaz et al. reported the sorption behavior of Evans Blue by hexamethylene diisocyanate (HMDI) crosslinked β-CD polymer,9 and Freundlich coefficient Kf was 0.57 which is close to the value of ASP adsorbed by β-CDP (Kf = 0.587) in this study. The Freundlich exponent 1/n has a dual significance: it is a measure of the percentage of high-affinity sites and also a measure of the heterogeneity in the system, and thus, is called the heterogeneity index.27 It typically varies from 0 to 1, with 1 being homogeneous and approaching zero being increasingly heterogeneous.27,28,31 In the present study, the values of 1/n measured for all the three polymers with ASP are above 1. This has been reported in the literature,8,32 and attributed to the fact that 1/n is both a measure of the ratio of high- to low-affinity sites and the heterogeneity of the binding sites, that suggests that the polymers that show a more favorable ratio of high- to low-affinity sites are increasingly heterogeneous.27
Sorption kinetics
The sorption kinetics of three pharmaceuticals to different CDPs are shown in Fig. 6. It could be seen that with the increase of the sorption time, the partitioning of pharmaceuticals to the sorbent gradually increased. As shown in Table 2, high sorption rates are observed at the beginning and more than 70% of the equilibrium amount is reached within 10 min for most of the CDPs except the adsorption of APAP to α-CDP (69.4%). For ASP and APAP, the sorption rates of different CDPs increased in the order: α-CDP < β-CDP < γ-CDP. This might be attributed to the limited size of ASP and APAP that are expected to be included into the CD cavities. In the case of LVF, γ-CDP also exhibits the highest sorption rate compared to the α- and β-analogues, but β-CDP shows the lowest sorption rate, which is different to that of ASP and APAP. This might be attributed to the molecular size of LVF that has a tri-membered ring too bulky to fit easily inside the CD cavities.33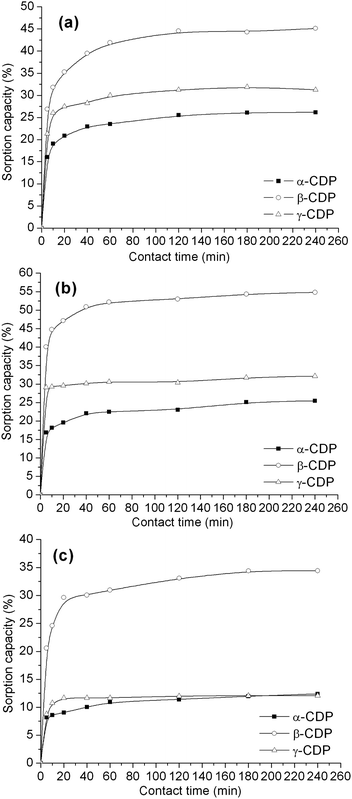 | ||
| Fig. 6 Kinetic sorption profiles of (a) LVF; (b) ASP; (c) APAP to α-CDP, β-CDP and γ-CDP. (CDP: 20 mg; pharmaceutical solution: 1.0 × 10−3 mol L−1, 2 mL.) | ||
| CDP | LVF | ASP | APAP | |||
|---|---|---|---|---|---|---|
| C 10/C0a (%) | SC b (%) | C 10/C0a (%) | SC b (%) | C 10/C0a (%) | SC b (%) | |
| a C 10/C0 is defined as the percent sorption to the equilibrium adsorbed amount within 10 min. b SC is the abbreviation of Sorption Capacity, defined as the percent amount of pharmaceuticals adsorbed to CDPs within 240 min. | ||||||
| α | 72.9 | 26.2 | 71.4 | 25.5 | 69.4 | 12.4 |
| β | 70.5 | 45.1 | 81.8 | 54.8 | 71.5 | 34.4 |
| γ | 83.4 | 31.3 | 91.3 | 32.1 | 89.3 | 12.1 |
The highest final sorption of β-CDP suggests that: (i) the cavity size of β-CD in the polymer may be suitable to include the ASP or APAP molecule, and consequently formed the most stable inclusion complex;30 (ii) the sorption mechanism is not only due to the formation of a CD/pharmaceutical inclusion complex, but also to the adsorption/diffusion in the CD polymer network. Concerning LVF, β-CDP also shows higher sorption capacity with regard to α-CDP and γ-CDP.
Conclusions
Three water insoluble CD-based polymers (CDPs) were prepared and used as sorbents for binding of three selected pharmaceuticals in aqueous solutions. The results indicated that the sorption of the pharmaceuticals on CDPs is consistent with the Freundlich model. β-CDP showed the highest sorption capacity for the tested pharmaceuticals. The interaction kinetics have been shown to be fast with more than 70% of the equilibrium amount was adsorbed within 10 min except the adsorption of APAP to α-CDP. This approach of such structure/property relationships is expected to be of interest for the design of new CD-based sorbents for the removal of organic contaminant from water.Acknowledgements
The financial support of the Swiss Commission for Technology and Innovation (CTI) is gratefully acknowledged. The authors would like to thank Dr Dirk Elend, Alessandro Cumbo and Helmut Fally for their assistance with HPLC, SEM and BET experiments, respectively.Notes and references
- P. J. Vikesland and K. R. Wigginton, Environ. Sci. Technol., 2010, 44, 3656–3669 CrossRef CAS.
- B. Sellergren and C. J. Allender, Adv. Drug Delivery Rev., 2005, 57, 1733–1741 CrossRef CAS.
- H. Asanuma, T. Akiyama, K. Kajiya, T. Hishiya and M. Komiyama, Anal. Chim. Acta, 2001, 435, 25–33 CrossRef CAS.
- J. W. Steed and J. L. Atwood, Supramolecular Chemistry, John Wiley & Sons Ltd., Chichester, 2009 Search PubMed.
- F. van de Manakker, T. Vermonden, C. F. van Nostrum and W. E. Hennink, Biomacromolecules, 2009, 10, 3157–3175 CrossRef CAS.
- P. Shahgaldian and U. Pieles, Sensors, 2006, 6, 593–615 CrossRef CAS.
- J. Szejtli, Chem. Rev., 1998, 98, 1743–1754 CrossRef CAS.
- S. Tang, L. Kong, J. Ou, Y. Liu, X. Li and H. Zou, J. Mol. Recognit., 2006, 19, 39–48 CrossRef CAS.
- E. Yilmaz, S. Memon and M. Yilmaz, J. Hazard. Mater., 2010, 174, 592–597 CrossRef CAS.
- L. Strimbu, J. Liu and A. E. Kaifer, Langmuir, 2003, 19, 483–485 CrossRef CAS.
- G. Crini, Bioresour. Technol., 2003, 90, 193–198 CrossRef CAS.
- A. Yilmaz, E. Yilmaz, M. Yilmaz and R. A. Bartsch, Dyes Pigm., 2007, 74, 54–59 CrossRef CAS.
- İ. Abay, A. Denizli, E. Bişkin and B. Salih, Chemosphere, 2005, 61, 1263–1272 CrossRef CAS.
- H. Yamasaki, Y. Makihata and K. Fukunaga, J. Chem. Technol. Biotechnol., 2006, 81, 1271–1276 CrossRef CAS.
- Z. Xu, L. Xu, D. Kuang, F. Zhang and J. Wang, Mater. Sci. Eng., C, 2008, 28, 1516–1521 CrossRef CAS.
- H. Yamasaki, Y. Makihata and K. Fukunaga, J. Chem. Technol. Biotechnol., 2008, 83, 991–997 CrossRef CAS.
- F. Zha, S. Li and Y. Chang, Carbohydr. Polym., 2008, 72, 456–461 CrossRef CAS.
- S. Murai, S. Imajo, Y. Takasu, K. Takahashi and K. Hattori, Environ. Sci. Technol., 1998, 32, 782–787 CrossRef CAS.
- J. C. Yu, Z.-T. Jiang, H.-Y. Liu, J. Yu and L. Zhang, Anal. Chim. Acta, 2003, 477, 93–101 CrossRef CAS.
- S. D. Mhlanga, B. B. Mamba, R. W. Krause and T. J. Malefetse, J. Chem. Technol. Biotechnol., 2007, 82, 382–388 CrossRef CAS.
- H. Liu, C. Liu, X. Yang, S. Zeng, Y. Xiong and W. Xu, Anal. Chim. Acta, 2008, 628, 87–94 CrossRef CAS.
- H. Liu, C. Liu, X. Yang, S. Zeng, Y. Xiong and W. Xu, J. Sep. Sci., 2008, 31, 3573–3580 CrossRef CAS.
- F. A. Caliman and M. Gavrilescu, Clean: Soil, Air, Water, 2009, 37, 277–303 Search PubMed.
- M. F. Rahman, E. K. Yanful and S. Y. Jasim, Desalination, 2009, 248, 578–585 CrossRef CAS.
- M. Whelehan, U. von Stockar and I. W. Marison, Water Res., 2010, 44, 2314–2324 CrossRef CAS.
- T. Hishiya, M. Shibata, M. Kakazu, H. Asanuma and M. Komiyama, Macromolecules, 1999, 32, 2265–2269 CrossRef CAS.
- M. Yan and O. Ramström, Molecularly Imprinted Materials: Science and Technology, Marcel Dekker, New York, 2005 Search PubMed.
- G. T. Rushton, C. L. Karns and K. D. Shimizu, Anal. Chim. Acta, 2005, 528, 107–113 CrossRef CAS.
- I. X. García-Zubiri, G. González-Gaitano and J. R. Isasi, J. Colloid Interface Sci., 2009, 337, 11–18 CrossRef CAS.
- K. Uekama, M. Otagiri, T. Irie, H. Seo and M. Tsuruoka, Int. J. Pharm., 1985, 23, 35–42 CrossRef CAS.
- R. J. Umpleby, S. C. Baxter, A. M. Rampey, G. T. Rushton, Y. Chen and K. D. Shimizu, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2004, 804, 141–149 CrossRef CAS.
- A. Romo, F. J. Peñas and J. R. Isasi, J. Colloid Interface Sci., 2004, 279, 55–60 CrossRef CAS.
- W. Tian, X. Fan, J. Kong, Y. Liu, T. Liu and Y. Huang, Polymer, 2010, 51, 2556–2564 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |