Synthesis of hydrogels via ring-opening metathesis polymerization: factors affecting gelation
Ahmad E.
Madkour
,
Joshua M.
Grolman
and
Gregory N.
Tew
*
Department of Polymer Science & Engineering, University of Massachusetts, 120 Governors Drive, Amherst, MA 01003, USA. E-mail: tew@mail.pse.umass.edu; Fax: +1-413-545-0082
First published on 1st November 2010
Abstract
Ring-opening metathesis polymerization (ROMP) was used to synthesize hydrogels via copolymerization of a diamine monomer 3 and a novel cross-linker 5 using Grubbs' third generation catalyst as initiator. Reactions were performed at two different monomer concentrations and at various initial molar ratios of cross-linker to initiator. At low monomer concentration, gelation occurred at initial cross-linker to initiator ratios of 1.5 and greater, which decreased to values of 1.05 and greater when increasing the monomer concentration. This result is in agreement with the Flory–Stockmayer theory. The gel yield and swelling properties were also found to be dependent on the cross-linker to initiator molar ratios. GPC data of the sol fractions showed quantitative consumption of both the monomer and the cross-linker. The molecular weight of the sol fraction was independent of the initiator concentration at constant cross-linker concentration of 2.5 mol%. Gels were found to swell up to 72 times their weight in 0.9% NaCl solution.
Introduction
Hydrogels and polyelectrolyte networks have received much attention because of their numerous applications in fields such as superabsorbent materials, soft contact lenses, tissue engineering, controlled drug delivery, and regenerative medicine.1–3 The most common method for preparing these networks is by the copolymerization of a hydrophilic or charged monomer with a cross-linker.4 Free-radical polymerization (FRP) has been widely used for constructing these networks because of its mild experimental conditions and the wide range of monomers that can be used. However, FRP has little control over the primary polymer chain network structure, which leads to microgel formation and a heterogeneous network structure.5 This is attributed to slow initiation, fast chain propagation, and the presence of termination reactions.5 At the beginning of the polymerization, individual chains with high molecular weight and a large number of pendant double bonds are formed in a short time. The relatively high dilution of chains in solution combined with slow diffusion and fast propagation rates leads to rapid intramolecular reactions between propagating chains and pendant double bonds. This generates various loops and densely cross-linked domains (microgels). These structural features then lead to the above mentioned heterogeneities.One method to improve the homogeneity of the network is to employ controlled radical polymerization (CRP) techniques, including atom transfer radical polymerization (ATRP),4–7 reversible addition fragmentation chain transfer (RAFT),8 and nitroxide-mediated polymerization.9,10 These approaches afford networks with more homogeneous structures because of their inverse reaction kinetics (fast initiation and slow chain growth).10,11 Armes and coworkers studied the copolymerization of 2-hydroxypropyl methacrylate with ethylene glycol dimethacrylate (EGDMA) as a cross-linker under ATRP conditions.12 Soluble branched copolymer (sol) was obtained when less than one branching agent (EGDMA) was incorporated per primary polymer chain. Higher levels of EGDMA (1.1 mol per primary chain) led to gelation. Similarly, the copolymerization of methyl acrylate and ethylene glycol diacrylate (EGDA) using ATRP led to gel formation at initial cross-linker to initiator molar ratios ([CL]o/[I]o) equal to or greater than 1.1.5 This shows that the reactions using ATRP closely follow the predictions of Flory and Stockmayer for gelation, which state that gelation occurs when the cross-linking index is unity.13–16
In the case of branched polymer synthesis using RAFT, several studies showed strong deviations from the Flory–Stockmayer predictions (up to 3.6 cross-linker units per polymer chain without forming gels).11,17–19 This may be attributed to a considerable degree of intramolecular cyclization rather than the desired intermolecular reaction. Intramolecular cyclization can be suppressed by conducting RAFT copolymerization at a relatively high monomer concentration, similar to that used in ATRP (i.e. ∼50% w/w instead of ∼20% w/v).20 Gelation by this RAFT formulation more closely conforms to the Flory–Stockmayer theory.
Ring-opening metathesis polymerization (ROMP) is another living/controlled polymerization method, and thus appeared to be a promising method to control the gelation process. ROMP has become a valuable tool for the preparation of a broad variety of materials. Among many types of ROMP catalyst systems, ruthenium-based complexes, particularly Grubbs' 1st to 3rd generation catalysts, are the most successful because they display high reactivity at room temperature and are relatively stable under ambient conditions.21,22 Furthermore, they are tolerant to functional groups including alcohols, acids and tertiary amines.23–25 The synthesis of ROMP networks is known, but mainly for hydrophobic monomers, and has been typically used to make thermosets.26,27 To our knowledge, there are only two previous reports of ROMP based hydrophilic networks. Hamilton and coworkers polymerized norbornene-dicarboxylic acid anhydride in the presence of an alcohol and a diol to obtain gels that could absorb up to 83 weight% water.28 Buchmeiser and coworkers polymerized norbornene-dicarboxylic acid anhydride in the presence of a cross-linker to form a polyanhydride network, which was then hydrolyzed to yield a polyelectrolyte network.29 We have previously shown that the ROMP polymerization of the oxa-norbornene diamine monomer 3 was living (Scheme 1), and that well-defined, high molecular weight polymers can be obtained with complete conversion within minutes.24 Dynamic light scattering experiments showed that these polymers were completely water-soluble, with negligible aggregation.
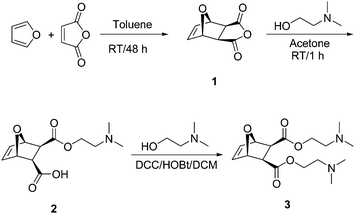 | ||
| Scheme 1 Synthesis of monomer 3. | ||
Here, we report the synthesis of polynorbornene-based hydrogels by polymerization of the diamine monomer 3 in the presence of the novel cross-linker 5via ROMP using Grubbs' third generation catalyst as initiator. The oxa-norbornyl moiety in both the monomer and the cross-linker was held constant, suggesting similar polymerization rates. The effect of monomer concentration and [CL]o/[I]o on gel formation, along with the gel yield and its swelling properties were explored. Our results confirm that reactions under ROMP conditions, at relatively high monomer concentration, closely follow the predictions of Flory and Stockmayer for network formation. Additionally, we found that our gels were able to absorb up to 72 times their weight of 0.9 wt% NaCl solutions.
Experimental
Instruments
1H NMR spectra were recorded on a 300 MHz Bruker Spectrospin 300. Gel permeation chromatography (GPC) in DMF/0.01 M LiCl was performed using a Polymer Laboratories PL-GPC50 instrument with two 5 µm mixed-D columns, a 5 µm guard column, and a Knauer RI detector. The system was calibrated against polystyrene standards, with toluene as the flow marker.Materials
Furan, maleic anhydride, N,N-dimethylaminoethanol, 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC), N,N′-dicyclohexylcarbodiimide (DCC), 1-hydroxybenzotriazole, anhydrous (HOBt), 1,6-hexanediol, ethyl vinyl ether and 4-dimethylaminopyridine (DMAP) were obtained from Sigma-Aldrich, Chem. Impex or Acros and used without further purification. THF (Fisher Scientific) was distilled over sodium and benzophenone under nitrogen before use. DCM (VWR) was distilled over CaH2 and purged with N2 before use. Poly(acrylic acid) partial sodium salt, particle size < 1000 µm (Aldrich). The third generation Grubbs' catalyst, G3 (dichloro-di(3-bromopyridino)-N,N′-dimesitylenoimidazolino-Ru![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHPh) was synthesized as described previously.30exo-7-Oxabicyclo[2.2.1]hept-5-ene-2,3-dicarboxylic anhydride 1 was synthesized according to literature procedure.24
CHPh) was synthesized as described previously.30exo-7-Oxabicyclo[2.2.1]hept-5-ene-2,3-dicarboxylic anhydride 1 was synthesized according to literature procedure.24
Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) to give a white precipitate which was filtered and crystallized from DCM/hexanes to give 15.6 g of pure product (yield = 61%). 1H NMR (300 MHz, CDCl3) δ = 1.23 (t, J = 7.15 Hz, 3H, CH3), 2.78 (d, J = 9.0 Hz, 1H), 2.87 (d, J = 9.0 Hz, 1H), 4.14 (q, J = 7.15 Hz, 2H), 5.24 (s, 1H), 5.29 (s, 1H), 6.45 (d, J = 6.7 Hz, 1H), 6.48 (d, J = 6.7 Hz, 1H), 10.9 (br s, 1H, COOH).
:1, v/v) to give a white precipitate which was filtered and crystallized from DCM/hexanes to give 15.6 g of pure product (yield = 61%). 1H NMR (300 MHz, CDCl3) δ = 1.23 (t, J = 7.15 Hz, 3H, CH3), 2.78 (d, J = 9.0 Hz, 1H), 2.87 (d, J = 9.0 Hz, 1H), 4.14 (q, J = 7.15 Hz, 2H), 5.24 (s, 1H), 5.29 (s, 1H), 6.45 (d, J = 6.7 Hz, 1H), 6.48 (d, J = 6.7 Hz, 1H), 10.9 (br s, 1H, COOH).
Cross-linker (5)
Compound 4 (5.0 g, 23.6 mmol), 1,6-hexanediol (0.93 g, 7.9 mmol) and DMAP (0.3 g, 2.46 mmol) were dissolved in dry DCM (50 mL) under N2. The resulting solution was then cooled to 0 °C, and 4.5 grams of EDC (4.5 g, 23.5 mmol) were added to the mixture in portions. The reaction mixture was then allowed to warm to RT while stirring and left for 12 h. The mixture was washed with 10% KHSO4 solution, sat. NaHCO3 solution, then brine. The resulting DCM solution was dried using anhydrous Na2SO4, filtered, and the solvent was evaporated. The resulting residue was purified using column chromatography (silica, hexanes/ethyl acetate) to afford 2.73 g of colorless oil (yield = 69%). 1H NMR (300 MHz, CDCl3) (mixture of 2 diastereomers) δ 1.28 (t, J = 7.16, 6H, CH3), 1.37–1.43 (m, 4H), 1.62–1.7 (m, 4H), 2.82 (s, 4H), 4.04–4.23 (m, 8H), 5.27 (m, 2H), 5.28 (m, 2H), 6.48 (br s, 4H). 13C NMR (75 MHz, CDCl3) δ 13.73, 25.11, 27.91, 46.38, 46.42, 60.52, 64.50, 80.04, 136.22, 171.01, 171.15. FAB HRMS found 507.2206, calculated 507.2230 [(M + H)+, M = C26H34O10].Gel synthesis
Certain amounts of monomer and cross-linker were placed in a 25 mL scintillation vial and dissolved in dry, degassed DCM. G3 catalyst was dissolved in DCM and added to the monomer/cross-linker solution. The mixture was kept at 30 °C for 1 h. Unreacted monomer, cross-linker and/or sol fraction were removed from the gel by repeated DCM extraction. DCM was replaced twice a day for seven days. Gels were dried under N2 flow then under vacuum for 24 hours.Gel swelling
The dried gel was swollen in RO water (500 mL) or in 0.9 wt% NaCl solution, and was left for 24 hours. The swollen gel was taken out, placed in a Buchner funnel fitted with a porous plastic film. The swollen gel was then weighed using a plastic weighing boat. The swelling capacity was then calculated using the following equation:| swelling capacity = [Wi − Wo]/Wo |
Results and discussion
Synthesis
Scheme 1 shows the modified synthesis of the previously reported diamine monomer 3.24 The exo-anhydride 1 was reacted with N,N-dimethylaminoethanol to form the monoester 2via a ring-opening reaction. Condensation of a second equivalent of N,N-dimethylaminoethanol with 2 using DCC as a coupling agent and HOBt afforded the diamine monomer 3. DCC was employed as a coupling agent because the urea byproduct could be easily removed from the reaction mixture by filtration. Although the reaction yield was almost quantitative as confirmed by 1H NMR spectroscopy of the crude product, the isolated yield of 3 was only ∼44% as the product was difficult to purify and trace amounts of HOBt impurities quenched the polymerization. Cross-linker 5 was synthesized by coupling 1,6-hexanediol with an excess of the monoester 4 using EDC as a coupling agent (Scheme 2). A typical polymerization leading to gel formation was performed by dissolving both monomer and cross-linker in DCM and adding Grubbs' third generation catalyst to the mixture under N2. The mixture was kept at 30 °C for one hour to ensure maximum monomer consumption. Gelation occurred within two minutes and no low molecular weight peaks that could be attributed to monomer or cross-linker were detected by GPC analysis, indicating monomer consumption was near quantitative.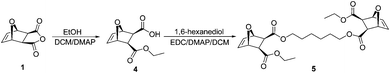 | ||
| Scheme 2 Synthesis of cross-linker 5. | ||
Effect of monomer and initiator concentration on gel formation
Several experimental parameters were expected to have an influence on gel formation and the resulting properties. Variables included the efficiency of the initiator, the reactivity of the cross-linker double bonds compared to that of monomer, the initial monomer concentration and the number of cross-linker units per polymer chain. In this study, we chose G3 as initiator because of its high initiation efficiency, and its tolerance to functional groups.21,24,31 We have shown previously that the polymerization of monomer 3 with G3 is living/controlled, resulting in polymers with low polydispersity (PDI = 1.05–1.08) and high conversion (99%) with good agreement between targeted and obtained polymer molecular weight.Initially, the gel synthesis was carried out under relatively low monomer concentration (0.51 mol L−1), and the cross-linker amount was varied from 1 to 4 mol% with respect to the monomer. For each of these cross-linker concentrations, we studied the effect of [CL]o/[I]o on gel formation, yield and swelling by holding monomer and cross-linker concentrations constant, while the initiator concentration was varied. The results of these studies are shown in Table 1 (entries 1–18). These data show that gel formation was dependent on [CL]o/[I]o, and that no gelation occurred with [CL]o/[I]o values of 1.4 or less at this low monomer concentration. This is consistent with other reported values of [CL]o/[I]o required for gelation. When copolymerizing 2-hydroxypropyl acrylate and EGDA by RAFT, no gelation occurred at [CL]o/[I]o values below 1.8.18 Similar observations were reported by Taton et al.,17 and Perrier and coworkers.11
| Entry | [M]o/mol L−1 | CLb (mol%) | [CL]o/[I]o | [M]o/[I]oc | Gelation or no gelation |
|---|---|---|---|---|---|
| a Experimental conditions: reactions were carried out in DCM at 30 °C for 60 minutes. b With respect to [M]o. c [M]o/[I]o is the initial monomer to initiator molar ratio. | |||||
| 1 | 0.51 | 1 | 0.90 | 88 | No gelation |
| 2 | 0.51 | 1 | 1.75 | 176 | Gelation |
| 3 | 0.51 | 2 | 0.90 | 45 | No gelation |
| 4 | 0.51 | 2 | 1.18 | 59 | No gelation |
| 5 | 0.51 | 2 | 1.75 | 88 | Gelation |
| 6 | 0.51 | 2 | 3.52 | 176 | Gelation |
| 7 | 0.51 | 3 | 1.34 | 45 | No gelation |
| 8 | 0.51 | 3 | 1.73 | 59 | Gelation |
| 9 | 0.51 | 3 | 2.61 | 88 | Gelation |
| 10 | 0.51 | 3 | 5.23 | 176 | Gelation |
| 11 | 0.51 | 4 | 1.3 | 35 | No gelation |
| 12 | 0.51 | 4 | 1.4 | 37 | No gelation |
| 13 | 0.51 | 4 | 1.5 | 40 | Gelation |
| 14 | 0.51 | 4 | 1.6 | 43 | Gelation |
| 15 | 0.51 | 4 | 1.7 | 45 | Gelation |
| 16 | 0.51 | 4 | 1.8 | 45 | Gelation |
| 17 | 0.51 | 4 | 2.29 | 59 | Gelation |
| 18 | 0.51 | 4 | 3.56 | 88 | Gelation |
| 19 | 1.28 | 4 | 0.95 | 25 | No gelation |
| 20 | 1.28 | 4 | 1.05 | 28 | Gelation |
| 21 | 1.28 | 4 | 1.1 | 29 | Gelation |
| 22 | 1.28 | 4 | 1.2 | 32 | Gelation |
| 23 | 1.28 | 4 | 1.3 | 35 | Gelation |
| 24 | 1.28 | 4 | 1.4 | 37 | Gelation |
| 25 | 1.28 | 4 | 1.5 | 40 | Gelation |
| 26 | 1.28 | 4 | 1.6 | 43 | Gelation |
According to the Flory–Stockmayer theory,13–16 gelation occurs when the cross-linking index (average number of cross-links per primary polymer chain in the system) is unity, i.e. on average, there are two branch points (cross-link points) on every polymer chain, assuming all polymerizable groups have the same reactivity, and the absence of intramolecular cyclization. For controlled polymerization, [CL]o/[I]o is equivalent to the number of cross-linkers per primary polymer chain (assuming that every initiator produces one primary polymer chain). In accordance with this, no gelation should be observed when [CL]o/[I]o is less than unity. Generally, experimental values of [CL]o/[I]o are slightly greater than the theoretical value of one, most likely because of undesirable side reactions such as intramolecular cyclization. It has been shown that the degree of intramolecular cyclization can be minimized by increasing the monomer concentration.20,32,33 In order to obtain critical [CL]o/[I]o values closer to the theoretical value of one, we attempted to reduce intramolecular cyclization by increasing the monomer concentration to 1.28 mol L−1. Gelation was examined at different [CL]o/[I]o values; cross-linker concentration was kept constant at 4 mol% while the initiator concentration was varied (Table 1, entries 19–26).
As mentioned, at low monomer concentration (0.51 mol L−1), no gelation occurred up to a [CL]o/[I]o value of 1.4. However, at the higher monomer concentration (1.28 mol L−1), gelation occurred at a [CL]o/[I]o value as low as 1.05. As expected, these conditions reduced the intramolecular cyclization reactions which were favored in dilute solution and had no cross-linking contribution to the gel network. This is in agreement with reports by Armes and coworkers on RAFT20 gelation and Gao et al. on networks via ATRP.33
Gel fraction and swelling properties
Two series of experiments were performed at 2.5 and 4 mol% cross-linker concentrations with varying [CL]o/[I]o values. For each series, monomer and cross-linker concentrations were kept constant while initiator concentration was varied. The gel fraction (yield) was measured at complete monomer conversion. Fig. 1 shows that the gel fraction increases with decreasing amount of initiator, suggesting that an increase in the molecular weight of the primary chain leads to less sol and more gel because polymers with higher molecular weights contain a larger number of pendent cross-linking units per chain. Thus, they have an increased probability of reacting with other chains, forming highly branched polymers, and ultimately the gel network.7 Higher initiator concentrations lead to shorter polymer chains with less cross-linking units per chain, so the probability of these chains reacting with each other and forming a network is reduced.7 Consequently, at high initiator concentrations, a larger fraction of low molecular weight branched polymers that are not part of a network was obtained. The molecular weight of the sol fraction was investigated by GPC (Fig. 2). As shown, the molecular weight of the sol fractions was almost constant (∼35 kDa for 2.5% cross-linker) irrespective of the initiator concentrations.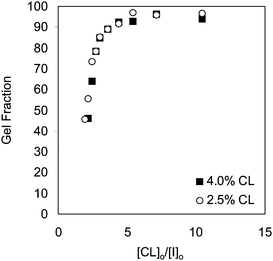 | ||
| Fig. 1 Effect of initial cross-linker to initiator molar ratio on gel fraction, at 2.5% (○) and 4% CL (■) molar ratios with respect to monomer. | ||
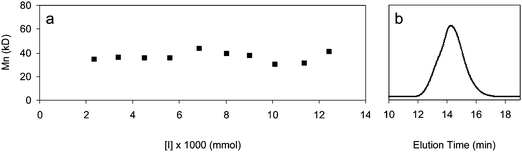 | ||
| Fig. 2 (a) Effect of the initial initiator concentration on the molecular weight of the sol fraction after complete monomer conversion at 2.5% cross-linker with respect to monomer. (b) Representative GPC trace (DMF, PS Standards) of sol fraction extracted from gel. | ||
It has been shown previously by dynamic light scattering that homopolymers from monomer 3 do not aggregate in aqueous solutions.24 Based on these results, we expected good swelling properties for our gels as these materials are evidently sufficiently hydrophilic.24 The swelling capacities of the gels in both 0.9 wt% NaCl solution and RO water were found to increase with increasing initiator concentration at constant monomer and cross-linker concentrations (Fig. 3). This is in line with the previous discussion of results in which higher initiator concentrations produced shorter chains with fewer cross-linking units, leading to loosely cross-linked networks with higher swelling capacities. Networks formed by ATRP show similar behavior.5 Gao et al. showed that the gel fraction of samples prepared from methyl acrylate and EGDA was ∼95% at a [CL]o/[I]o value of 1.1, and only 45% when the ratio was ∼10. In addition, an increase in initiator concentration resulted in an increase in swelling capacity of the gel, from ∼2 to 40 g per g in THF.5
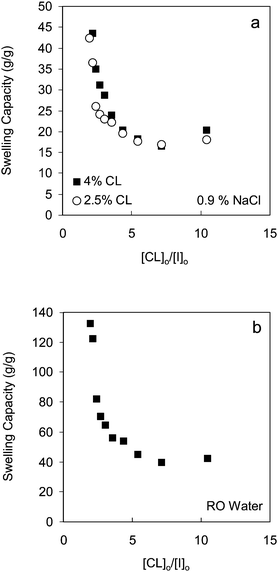 | ||
| Fig. 3 Effect of initial cross-linker to initiator molar ratios on gel swelling capacities: (a) 2.5% cross-linker (○) and 4% cross-linker (■) in 0.9% NaCl solution and (b) 2.5% cross-linker in RO water respectively. | ||
Influence of cross-linker percentage on swelling
The swelling capacity of a gel is known to be affected by the cross-linker content.13 The swelling behavior of our gels as a function of mol percentage of cross-linker was studied at two different initial monomer to initiator molar ratios. As shown in Fig. 4, the swelling capacities of the gels decreased with increasing cross-linker content, which is to be expected, as a high cross-linking density decreases the molecular weight between the cross-links and results in a rigid structure that can not expand to hold as much liquid as a more loosely cross-linked network. These gels can absorb an amount of 0.9 wt% NaCl solution that is up to 72 times their own weight.![Effect of cross-linker percentage on gel swelling capacity in 0.9% NaCl solution at two different initial monomer to initiator concentration ([M]o/[I]o).](/image/article/2011/PY/c0py00151a/c0py00151a-f4.gif) | ||
| Fig. 4 Effect of cross-linker percentage on gel swelling capacity in 0.9% NaCl solution at two different initial monomer to initiator concentration ([M]o/[I]o). | ||
pKa measurments
The swelling ability of polyelectrolyte gels is due to a combination of the repulsive electrostatic interactions between the charged groups on the polymer chains and the osmotic pressure between the interior and exterior of the gel due to the counterions inside the network.13,34–38 The pKa value of the polymer will determine the extent of the amine group protonation at a given pH, and accordingly, the amount of charge on the chain. For our system, the monomer pKa value was found to be 8.13, whereas that of the polymer was 8.35 (Fig. 5). These measurements indicated that incorporation into a macromolecule did not significantly affect the ionization ability of the tertiary amino groups. At a pH of 7, according to these data, more than 50% of the tertiary amino groups of the polymer are protonated. This means that at neutral pH the polymer is charged, leading to its high degree of swelling in salt water.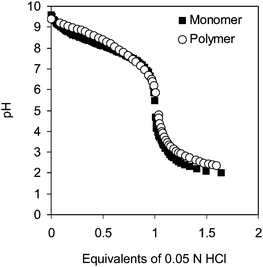 | ||
| Fig. 5 Titration curves for monomer 3 and corresponding polymer obtained by titrating aqueous solution of the polymer with 0.05 N HCl. | ||
Conclusions
We used ring-opening metathesis polymerization to synthesize a series of norbornene-based hydrogels through the copolymerization of a diamine monomer and a novel cross-linker using Grubbs' third generation catalyst as an initiator. The gelation was found to be dependent on the initial cross-linker to initiator molar ratio. At higher monomer concentration (1.28 g L−1), gel formation occurred at ratios of 1.05 and higher, which is in agreement with Flory–Stockmayer theory. This behavior is due to the preferential intermolecular branching at high monomer concentration, as opposed to intramolecular loop formation, which occurs at lower monomer concentration (0.51 g L−1). Varying the cross-linker to initiator molar ratio at constant monomer concentrations was found to greatly affect gel yields and swelling properties. While the gel fraction was found to increase with decreasing initiator amount, the swelling capacity decreased. GPC analysis of the sol fractions revealed that molecular weight of the sol fraction is independent of the initiator concentration at the constant cross-linker concentration studied. We found that our gels have a swelling capacity up to 72 times their weight in 0.9 wt% NaCl solutions, compared to 45 times for superabsorbent gels based on polyacrylic acid.39 Studies on the homogeneity of the gels at the micro- and nanoscale levels as well as mechanical characterization are currently under way and will be reported in due course.Acknowledgements
The authors gratefully acknowledge support for this work from ONR (N00014-07-1-0520). This work utilized facilities supported in part by Center for Hierarchical Manufacturing (CMMI 0531171). Melissa Lackey and Dr Karen Lienkamp are gratefully acknowledged for their helpful discussions.Notes and references
- A. S. Hoffman, Adv. Drug Delivery Rev., 2002, 54, 3–12 CrossRef CAS.
- J. L. Drury and D. J. Mooney, Biomaterials, 2003, 24, 4337–4351 CrossRef CAS.
- J. Kopecek, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5929–5946 CrossRef CAS.
- C. F. Jiang, Y. Q. Shen, S. P. Zhu and D. Hunkeler, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 3780–3788 CrossRef CAS.
- H. Gao, K. Min and K. Matyjaszewski, Macromolecules, 2007, 40, 7763–7770 CrossRef CAS.
- H. Gao, A. Miasnikova and K. Matyjaszewski, Macromolecules, 2008, 41, 7843–7849 CrossRef CAS.
- W. Li, H. Gao and K. Matyjaszewski, Macromolecules, 2009, 42, 927–932 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 379–410 CrossRef CAS.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661–3688 CrossRef CAS.
- N. Ide and T. Fukuda, Macromolecules, 1999, 32, 95–99 CrossRef CAS.
- B. L. Liu, A. Kazlauciunas, J. T. Guthrie and S. Perrier, Macromolecules, 2005, 38, 2131–2136 CrossRef.
- I. Bannister, N. C. Billingham, S. P. Armes, S. P. Rannard and P. Findlay, Macromolecules, 2006, 39, 7483–7492 CrossRef CAS.
- P. J. Flory, Principles of Polymer Chemistry, 1953 Search PubMed.
- P. J. Flory, J. Am. Chem. Soc., 1941, 63, 3083–3090 CrossRef CAS.
- W. H. Stockmayer, J. Chem. Phys., 1943, 11, 45–55 CrossRef CAS.
- W. H. Stockmayer, J. Chem. Phys., 1944, 12, 125–131 CrossRef CAS.
- D. Taton, J. F. Baussard, L. Dupayage, J. Poly, Y. Gnanou, V. Ponsinet, M. Destarac, C. Mignaud and C. Pitois, Chem. Commun., 2006, 1953–1955 RSC.
- C. D. Vo, J. Rosselgong, S. P. Armes and N. C. Billingham, Macromolecules, 2007, 40, 7119–7125 CrossRef CAS.
- Y. T. Li and S. P. Armes, Macromolecules, 2009, 42, 939–945 CrossRef CAS.
- J. Rosselgong, S. P. Armes, W. Barton and D. Price, Macromolecules, 2009, 42, 5919–5924 CrossRef CAS.
- C. Slugovc, Macromol. Rapid Commun., 2004, 25, 1283–1297 CrossRef CAS.
- S. Hilf and A. F. M. Kilbinger, Nat. Chem., 2009, 1, 537–546 CrossRef CAS.
- K. Lienkamp, C. F. Kins, S. F. Alfred, A. E. Madkour and G. N. Tew, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 1266–1273 CrossRef CAS.
- S. F. Alfred, K. Lienkamp, A. E. Madkour and G. N. Tew, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6672–6676 CrossRef CAS.
- S. Colak and G. N. Tew, Macromolecules, 2008, 41, 8436–8440 CrossRef CAS.
- X. Sheng, J. K. Lee and M. R. Kessler, Polymer, 2009, 50, 1264–1269 CrossRef CAS.
- X. Sheng, M. R. Kessler and J. K. Lee, J. Therm. Anal. Calorim., 2007, 89, 459–464 CrossRef CAS.
- J. G. Hamilton, E. E. Law and J. J. Rooney, J. Mol. Catal. A: Chem., 1997, 115, 1–9 CrossRef CAS.
- M. R. Buchmeiser, N. Atzl and G. K. Bonn, J. Am. Chem. Soc., 1997, 119, 9166–9174 CrossRef CAS.
- J. A. Love, J. P. Morgan, T. M. Trnka and R. H. Grubbs, Angew. Chem., Int. Ed., 2002, 41, 4035–4037 CrossRef CAS.
- C. W. Bielawski and R. H. Grubbs, Prog. Polym. Sci., 2007, 32, 1–29 CrossRef CAS.
- Y. T. Li, A. J. Ryan and S. P. Armes, Macromolecules, 2008, 41, 5577–5581 CrossRef CAS.
- H. F. Gao, W. W. Li and K. Matyjaszewski, Macromolecules, 2008, 41, 2335–2340 CrossRef CAS.
- A. R. Khokhlov, S. G. Starodubtzev and V. V. Vasilevskaya, Adv. Polym. Sci., 1993, 109, 123–175 CAS.
- A. Katchalsky and I. Michaeli, J. Polym. Sci., 1955, 15, 69–86 CrossRef CAS.
- J. Wilder and T. A. Vilgis, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1998, 57, 6865–6874 CrossRef CAS.
- T. A. Vilgis and J. Wilder, Comput. Theor. Polym. Sci., 1998, 8, 61–73 CrossRef CAS.
- H. Frusawa and R. Hayakawa, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1998, 58, 6145–6154 CrossRef CAS.
- According to Sigma-Aldrich catalog, superabsorbent poly(acrylic acid) partial sodium salt particles were reported to absorb ∼45 g of 1% saline solution per g. Using our experimental condition, these particles absorb ∼40 g of 0.9% saline solution per g.
| This journal is © The Royal Society of Chemistry 2011 |