Visualizing the efficiency of rapid modular block copolymer construction†
Andrew J.
Inglis
and
Christopher
Barner-Kowollik
*
Preparative Macromolecular Chemistry, Institut für Technische Chemie und Polymerchemie, Karlsruhe Institute of Technology (KIT), Engesserstr. 18, 76128, Karlsruhe, Germany. E-mail: christopher.barner-kowollik@kit.edu
First published on 23rd August 2010
Abstract
The quantification of the efficient ultra-rapid modular synthesis of block copolymersviatwo-dimensional chromatography and a comparably accurate, low-cost deconvolution of size exclusion chromatography traces is reported. Cyclopentadienyl-capped poly(methyl methacrylate) (PMMA, 97% functionality) and a series of pyridin-2-yldithioformate-capped poly(isobornyl acrylate)s (PiBoA, 93–95% functionality) were synthesiszed via atom transfer radical polymerization (ATRP) and reversible addition fragmentation chain transfer (RAFT) polymerization respectively. The corresponding block copolymers were generated by simply stirring a chloroform solution of PMMA, PiBoA and acidic catalyst for 10 min at ambient temperature. The crude block copolymer mixtures were directly analyzed by size exclusion chromatography (SEC), liquid adsorption chromatography at critical conditions (LACCC) and 2D LACCC-SEC to visualize the efficiency of the ultra rapid conjugation reaction. The quantitative analysis via the 2D LACCC-SEC yielded the composition of the block copolymer mixtures which were in excellent agreement with predicted values, thus indicating quantitative conjugation efficiency. In all cases, the crude block copolymer mixtures contained at least 94 wt% block copolymer. Furthermore, a low-cost deconvolution method that may be applied to conventional SEC traces was found to provide comparable composition data.
Introduction
Despite their relative simplicity, block copolymers are highly sought after materials due to their ability to form well-defined micro-domains in both solution and in bulk.1 Such a property makes these materials highly useful in a wide variety of fields, including nanolithography,2–4 photonics,5 controlled drug delivery6 and nanotechnology in general. However, such ‘high-demand’ applications are substantially varied in their requirements, thus a rather broad range of high purity block copolymers are in need of being readily accessible.A versatile technique by which block copolymers may be synthesized is by modular construction. This technique is largely centered on the application of highly efficient organic chemical transformations to polymer chains synthesized by, primarily, controlled free radical polymerization (CRP). Techniques such as atom transfer radical polymerization (ATRP)7,8 and reversible addition fragmentation chain transfer (RAFT)9,10polymerization are very well suited to the generation of well-defined polymeric architectures that bear one or a multitude of synthetic handles, which may be effectively manipulated to achieve larger structures through conjugation. While anionic polymerization remains the dominant method of producing block copolymers (either by sequential polymerization or coupling reactions), the variety of monomers that may be polymerized is not as vast as what can be polymerized by CRP.11–13
While the transfer of synthetic organic chemistry techniques to polymer science has been a highly successful enterprise, the use of the associated purification techniques (e.g.column chromatography, distillation, and recrystallization) has not been possible. As such, every effort had been made to directly synthesize high purity block copolymers14–18 by employing only the most efficient chemical transformations that are on offer (often categorized as click chemistry).19 However, characterization of such efficiency is usually glossed over in conventional size exclusion chromatography (SEC) analyses and is not reliably quantified.
Liquid adsorption chromatography at critical conditions (LACCC) can be a very effective technique whereby macromolecules are separated by their chemical heterogeneity rather than their size.20–29 Thus, in the analysis of block copolymers (A-b-B) in which critical conditions are applied for the A segment, the elution behavior of the block will be solely dependent upon the B segment. Thus the A segment may be considered to be chromatographically invisible. However, some publications question and even challenge the validity of this assumption.30–32 This phenomenon may be advantageously used in establishing the efficiency of modular block copolymer construction where the conjugation of two homopolymers of significantly disparate molecular weight cannot convincingly be established by conventional SEC alone. By transferring eluates from the LACCC dimension to an SEC system (generically referred to as two-dimensional or 2D chromatography),24 a 2D-chromatogram (a two-dimensional projection of a three-dimensional surface) is thus obtained that provides information on both the chemical heterogeneity and size of the synthesized block copolymers. Critical conditions are strongly related to the polymer system under investigation. As such, for every different polymer that is in need of being analyzed by such a technique, the critical conditions must be individually determined.
Such an analysis has been effectively utilized by Matyjaszewski in establishing the efficiency of linear and star shaped block copolymer formation through a chain extension approach with various incarnations of ATRP.33–35 Furthermore, Falkenhagen and Müller et al. used the technique to characterize block copolymers formed by sequential living anionic polymerization.36 Pasch et al., in addition to the study of block copolymers formed by living anionic polymerization, explored the generation of poly(styrene)-block-poly(isoprene) copolymers by coupling separately synthesized poly(styrene) and poly(isoprene), also using anionic chemistry.37 Although the coupling process required three days, it nevertheless provided the inspiration to evaluate the efficiency of more rapid and versatile conjugation techniques.
Herein, we present a comprehensive assessment of the efficiency of formation and the resulting purity of block copolymers synthesized via an ultra rapid modular approach (specifically RAFT-HDA chemistry),17,18,38–43 as depicted in Scheme 1. Initially, cyclopentadienyl functional poly(methyl methacrylate) (PMMA 1) and a series of pyridin-2-yldithioformate functional poly(isobornyl acrylate)s (PiBoA 2, 3 and 4) were synthesized viaATRP and RAFT polymerization respectively and their end-group functionalities determined viaelectrospray ionization mass spectrometry (ESI-MS) and 1H-NMR spectroscopy. This allowed for an initial prediction of the expected compositions of the crude block copolymer mixtures working under the assumption of quantitative conjugation. After a reaction time of 10 min at ambient temperature, the crude block copolymer reaction mixtures were directly analyzed via conventional size exclusion chromatography (SEC), liquid adsorption chromatography at critical conditions (LACCC) and 2D LACCC-SEC to assess their purity. Finally, we will show how a simple deconvolution of the SEC traces of the crude block copolymer mixtures can be used to also achieve reliable composition data.
 | ||
| Scheme 1 Schematic overview of the conjugation of polymer chains via their respective chain-end functionalities. | ||
Experimental section
Materials
Benzyl pyridin-2-yldithioformate was synthesized according to the literature.44,45Methyl methacrylate (Acros) and isobornyl acrylate technical grade (Aldrich) were passed through a column of basic alumina (Acros) prior to use and stored at −19 °C. 2,2′-Azobis(2-methylpropionitrile) (AIBN, Sigma-Aldrich) was recrystallized twice from methanol prior to use and stored at −19 °C. Copper(I) bromide (Fluka) was purified by sequential washing with sulfurous acid, acetic acid and ethanol, followed by drying under reduced pressure prior to use. 2,2′-Bipyridyl (bpy, ≥99%, Sigma-Aldrich), copper(II) bromide (≥99%, Fluka), methyl 2-bromo-2-methylpropanoate (MBMP, ≥99%, Aldrich) and trifluoroacetic acid (TFA, Aldrich) were used as received.RAFT polymerization of isobornyl acrylate
A mixture of isobornyl acrylate, benzyl pyridin-2-yldithioformate and AIBN was deoxygenated by purging with nitrogen for 40 min. The polymerization reaction was performed at 60 °C for 24 h. The reaction was stopped by chilling in an ice bath and exposure to oxygen. The resulting polymers were isolated by two-fold precipitation in cold methanol. The specifics for each of the three polymerizations are as follows: PiBoA 2—[M]o![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[RAFT]o:[AIBN]o = 230:1:0.2, Mn = 4600 g mol−1, conversion = 10%; PiBoA 3—[M]o:[RAFT]o:[AIBN]o = 400:1:0.2, Mn = 8900 g mol−1, conversion = 10.5%; and PiBoA 4—[M]o:[RAFT]o:[AIBN]o = 650:1:0.2, Mn = 32000 g mol−1, conversion = 22%.
:[RAFT]o:[AIBN]o = 230:1:0.2, Mn = 4600 g mol−1, conversion = 10%; PiBoA 3—[M]o:[RAFT]o:[AIBN]o = 400:1:0.2, Mn = 8900 g mol−1, conversion = 10.5%; and PiBoA 4—[M]o:[RAFT]o:[AIBN]o = 650:1:0.2, Mn = 32000 g mol−1, conversion = 22%.
Preparation of PMMA 1
To a dried Schlenk tube were added copper(I) bromide, copper(II) bromide and bpy which was then sealed with a rubber septum, evacuated and backfilled with nitrogen. To another Schlenk tube were added methyl methacrylate (MMA) and acetone (50:50) and the tube sealed with a rubber septum. The monomer solution was then deoxygenated by three consecutive freeze–pump–thaw cycles and subsequently transferred to the first Schlenk tubevia cannula. The tube was sealed under a nitrogen atmosphere and placed in a thermostatted oil bath held at 50 °C. After the polymerization mixture reached the desired temperature, MBMP was added. The initial ratio of [MMA]:[MBMP]:[CuIBr]:[CuIIBr]:[bpy] was 50:2:0.105:0.0125:0.25. The polymerization was stopped by cooling the mixture in an ice-bath and exposure to oxygen. The mixture was passed through a short column of neutral alumina to remove the copper catalyst. Poly(methyl methacrylate) was isolated by two-fold precipitation in cold hexane. A solution of bromide terminated polymer (0.18 mmol), tributylphosphine (0.36 mmol, 2 eq.) and sodium iodide (1.08 mmol, 6 eq.) in anhydrous THF (2.0 mL) was prepared under a nitrogen atmosphere. Separately, a stock solution of NiCp2 in anhydrous THF (0.18 mol L−1) was prepared under a nitrogen atmosphere. The NiCp2 solution (2.0 mL, 4 eq.) was then added to the polymer solution and allowed to stir overnight at ambient temperature. At the end of the reaction, the mixture was passed through a short column of basic alumina to remove the precipitated nickel(II) bromide and the polymer recovered by precipitation. The resulting polymer was then dissolved in chloroform and washed three times with distilled water. The Cp-capped polymer was then isolated by precipitation from the chloroform solution into cold hexane. GPC (THF): Mn = 3200 g mol−1, PDI = 1.10.
Conjugation reactions-typical procedure
PMMA 1 (10 µmol) and PiBoA precursor were dissolved in chloroform (200 µL) such that an equimolar solution (based upon functional polymer) was obtained. For example, PMMA 1 (10 µmol, 97% functionality) was mixed with PiBoA 2 (10.2 µmol, 95% functionality). After the addition of TFA (1.5 eq.), the solution was agitated by shaking for 10 min at ambient temperature, after which the solvent was removed under reduced pressure to yield crude block copolymer mixtures, which were then directly analyzed without any purification.Measurements
Details concerning the analysis of the synthesized polymersviaLACCC and 2D LACCC-SEC are outlined in the following. Descriptions of other analytical techniques such as 1H-NMR, SEC and ESI-MS may be found in the ESI†.:MeOH (68:32 v/v%) eluent was pumped at a flow rate of 0.08 mL min−1. An evaporative light scattering detector (ELSD 400, SofTA Corporation) operating under an argon pressure of 47.6 psi and a spray chamber temperature of 50 °C was used. ELSD is the most bias-free (mass sensitive) detector when compared to RI or even UV detectors when analyzing block copolymers in terms of detecting both segments of the block with equal weighting. Samples were prepared in the above described eluent at a polymer concentration of 2 mg mL−1 (concentrations were systematically varied to obtain optimal results). Data acquisition and processing were accomplished with the PSS WinGPC Unity software package from Polymer Standards Service (PSS, Mainz, Germany).
SEC deconvolution
The SEC traces from the system outlined in the ESI† were deconvoluted using the Gaussian function of the Peak Fit program (version 4.12 from SeaSolve Software Inc.). The program uses a Gaussian response function with a Fourier deconvolution and filtering algorithm. The algorithm is run iteratively until the correlation coefficient of the fitted data is minimized (via the least squares method). It is necessary to use the retention times of the polymer precursors (which have been independently measured) to constrain fitted peaks to the corresponding values. The three placed peaks are then iteratively varied in their intensities and area such that the result of their convolution fits to the raw experimental data of the block copolymer.Results and discussion
In characterizing the success of block copolymer formation through a modular approach, it is of paramount importance to first determine the starting chain-end functionalities of the respective homopolymer precursors. A substantially versatile range of such polymeric building blocks may be (and has been) synthesized by controlled radical polymerization processes.48 Despite the highly effective level of control in terms of molecular weight and chain-end functionality offered by such processes as ATRP7,8 and RAFT9,10 polymerization, it is nevertheless impossible to achieve truly quantitatively functional polymer chains.ATRP and RAFT polymerization are often erroneously labeled as living free radical polymerization, however, the fact of the matter is that in both of these processes, various ill-desired termination, transfer and elimination reactions are not completely suppressed, thus a certain (albeit low) proportion of the total polymer that is recovered will be ‘dead polymer’, that is polymer chains that do not bear the targeted functionality. Such species usually cannot be preparatively separated from functional polymer and are thus carried through subsequent chemical transformations. Therefore, when it comes to the conjugation of two polymer chains via their respective chain-ends, the subsequent SEC analysis will ideally show the appropriate shift of the molecular weight distribution to higher molecular weights along with some additional tailing or even a pronounced hump towards lower molecular weight.49 Without knowing the starting chain-end fidelity, one cannot be certain which proportion of the SEC tailing is attributable to remaining ‘dead’ polymer chains, since a non-quantitative conjugation process would also lead to such an observation. Of course, other techniques such as NMR and FT-IR spectroscopy can be used to monitor the change in chemical functionality from the starting homopolymers to the final conjugate. Yet such analyses are typically dependent upon signals from small functional groups on the ends of long polymer chains. The higher the molecular weight of the polymers utilized become, the signal-to-noise ratio of the signals of interest becomes smaller, thus (of course after a certain limit) decreasing the accuracy of any quantitative analysis that may be performed. UV detection, on the other hand, is very sensitive and non-chain length dependent. However, this detection method is restricted to those species that absorb UV light and cannot therefore be applied to all compounds synthesized in the present investigation.
In the present circumstance, PMMA 1 was prepared viaATRP and the bromide end-group subsequently and directly transformed into a cyclopentadienyl moiety via our facile and previously reported procedure using nickelocene.50 Typically, the chain-end functionality of such polymers is determined via a 1H-NMR spectroscopic analysis in which the integrals of signals arising from end-group protons are compared. Due to the nature of PMMA 1, such an analysis cannot be reliably performed due to the lack of end-group protons that do not have overlapping signals with those that form part of the polymer backbone (Fig. S1†). As such, PMMA 1 was determined to contain 3 ± 0.60 wt% dead chains via quantitative integration of its mass spectrum (recorded viaelectrospray ionization mass spectrometry (ESI-MS)) following a previously reported procedure (Fig. S2†).51 In this technique, only the singly charged species were used for the integration. It is important to note that ionization occurs along the polymer backbone, a process that is not significantly influenced by the nature of the polymer end-group.51 Furthermore, it has been reported that the mole-fraction determination of ESI-MS data in (meth)acrylate-type systems is most likely systematically and largely independent of chain length.52 As reported, the error associated with this technique was estimated to be <20%. Thus, for the present investigation the maximum error of 20% was taken as a conservative estimate. Concurrently, a series of poly(isobornyl acrylate)s (PiBoA 2, 3 and 4) were synthesized viaRAFT polymerization mediated by pyridin-2-yldithioformate. Such a system produces polymer chains bearing the electron deficient dithioester end-group that is required for ultra-fast hetero Diels–Alder (HDA) cycloadditions.17,18 The molecular weights and functionalities (as determined by 1H NMR spectroscopy) of these polymers are presented in Table 1 (1H-NMR spectra of all homopolymers are included in the ESI†). As the molecular weight of a polymer increases, so does the error involved in determining end-group functionality via integration of its NMR spectrum. The two low molecular weight PiBoA's (2 and 3), a good signal-to-noise ratio is observed and, based upon multiple analyses (i.e. re-integration of the spectra on 5 occasions), the reproducibility of the determination was estimated to contain an error of ±5% (based on the variations of the repeated integration result). The NMR spectrum of the higher molecular weight PiBoA (4) was observed to have significantly lower signal-to-noise ratio and the reproducibility of its analysis by integration was estimated to have an error of 15% (also obtained by repeated integration). The 1H-NMR spectra were recorded with 128 scans (for the spectrometer details refer to the Experimental section).
| Polymer | M n,SEC/g mol−1 | PDI | Fraction of dead polymer chains (%) |
|---|---|---|---|
| a Determined via quantitative integration of the mass spectrum recorded by ESI-MS. b Determined via1H-NMR spectroscopy. | |||
| PMMA 1 | 3200 | 1.10 | 3.0 ± 0.60a |
| PiBoA 2 | 4600 | 1.09 | 5.0 ± 0.25b |
| PiBoA 3 | 8900 | 1.24 | 7.0 ± 0.35b |
| PiBoA 4 | 32000 |
1.08 | 6.0 ± 0.90b |
With highly functional homopolymers in hand, synthesis of the corresponding block copolymers was simply achieved by mixing the required components in a stoichiometric ratio (with respect to their available functional end-groups) with trifluoroacetic acid in a chloroform solution. After 10 min at ambient temperature, the solvent was removed in vacuo, yielding the targeted block copolymersPMMA-b-PiBoA 5, 6 and 7. It should be stressed at this point that no purification was performed on the crude reaction mixtures in order to avoid possible fractionation of the block copolymer from homopolymer precursors, thus the efficiency of the conjugations could truly be established.
Keeping in line with the most widely used technique in characterizing block copolymer formation, conventional SEC was employed to initially analyse the formed copolymers. The left hand side of Fig. 1 shows an overlay of the SEC traces of the various homopolymer precursors and of the respective block copolymers. In all cases, a clear shift to lower retention times may be observed, providing a qualitative indication of successful block formation. A comparison of the SEC determined molecular weights of the generated block copolymers and the predicted values (Table 2) also confirms the success of the conjugation. It may also be observed that a slight degree of tailing towards higher retention times is present in all of the examples, indicating the presence of seemingly unreacted starting materials. Now, upon first glance, it may seem that the conjugation reaction did not proceed quantitatively, as is the general interpretation of such results. However, as discussed previously, a small proportion of the starting homopolymers are not able to undergo the conjugation reaction for the simple reason that they do not carry the required dithioester or cyclopentadienyl functionality. Thus, a slight degree of tailing is expected. It is therefore necessary to determine whether such tailing is solely due to this dead polymeric material or if the conjugation reaction is indeed non-quantitative. Another limitation to the sole use of SEC to characterize the modular formation of block copolymers arises when there is a large difference in the molecular weight (or more precisely the hydrodynamic volume) of the two precursor homopolymers. Upon close inspection of the SEC section of Fig. 1, it is apparent that as the molecular weight of the PiBoA increases, the observable shift in retention time of the formed block copolymer becomes smaller. Taking a purely mathematical perspective on the convolution of two Gaussian-type distributions, the greatest shift will be observed when both starting components are very close to one another, if not identical. It is therefore highly useful in this case to separate the block copolymers from the PiBoA homopolymersvia their chemical composition in addition to their hydrodynamic volume. For this, an LACCC analysis was performed in which the critical conditions for PiBoA were utilized.
 | ||
| Fig. 1 SEC characterization (left) and LACCC characterization under the critical conditions of PiBoA (right) of the various homopolymers and block copolymers synthesized in this study. The molecular weights and degree of functionalization of these polymers are listed in Table 1. Note that the slightly non-Gaussian shape of some LACCC curves occurred at different analyte concentrations and could not be suppressed. However, the shape is of limited consequence as the analyte is transferred into the second dimension. | ||
Under the critical conditions for a specific polymer, the enthalpic and entropic interactions of that polymer with the stationary phase of the chromatographic column compensate each other, resulting in polymer chains eluting according to their chemical heterogeneity and independently of their size. Consequently, under the critical conditions of PiBoA, the elution behaviour of the block copolymers 5, 6 and 7 should differ from that of the corresponding PiBoA homopolymers. In reality, the compensating enthalpic and entropic effects can be less than perfect, resulting in the analyte eluting either slightly in size exclusion mode or in adsorption mode.
The right hand side of Fig. 1 shows the results of a LACCC analysis of all polymers and block copolymers synthesized under the critical conditions of PiBoA. Inspection of the LACCC section of Fig. 1 reveals: (1) PiBoA 2, 3, and 4 appear at a similar elution volume, thus confirming that the analysis has indeed been performed under the appropriate critical conditions; (2) PMMA 1 appears at a different elution volume to the PiBoA homopolymers, thus establishing the expectation that the block copolymers should also have a significantly different elution behaviour; and (3) the block copolymers 5, 6 and 7 do indeed display a substantially different elution behaviour compared to the PiBoA homopolymers, thus allowing for the presence of unreacted PiBoA homopolymer to be observed in the crude reaction mixtures. It should be noted that the HPLC column used for the LACCC analysis was thoroughly flushed with pure THF after each measurement. Upon monitoring the detector output during this process, no pronounced signals could be observed, indicating that no residual polymer was retained on the column. A cautionary note concerning the evaluation of data from ELSD is centred on its potentially—albeit unlikely—non-proportional response with regard to different polymer segments (see also below).
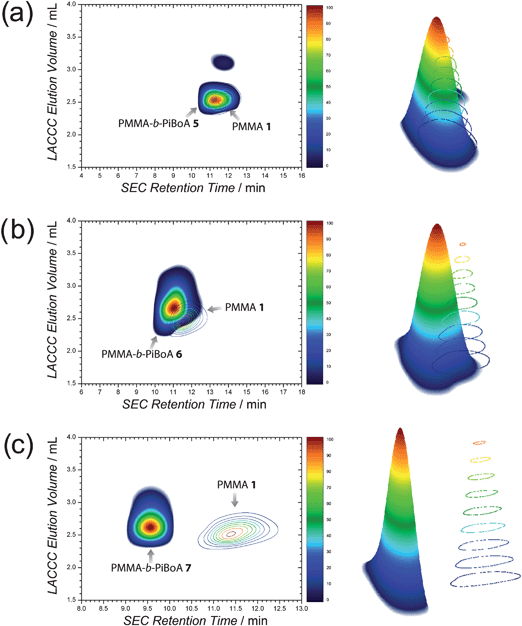
Upon combining the techniques of LACCC and SEC, contour plots of the synthesized block copolymers may be generated that provide information on both its chemical composition and size. Such a characterization of block copolymers has been reported in the literature;33–37,47 however, the present article provides the first comprehensive analysis of the efficiency and purity of block copolymers formed via a rapid modular pathway, of which 2D LACCC-SEC is but one element. Furthermore, this analysis enables the generation of a three-dimensional rendering of the block copolymers, thus aiding in the visualization of the efficiency of the modular conjugation.
Fig. 2 depicts the 2D chromatograms of the various block copolymers overlayed with those of the respective PiBoA homopolymers from which they were synthesized. The main 2D contour of Fig. 2a appearing at a LACCC elution volume of around 2.6 mL represents the PMMA-b-PiBoA 5. Associated with this contour is a much smaller contour that appears at a LACCC elution volume of around 3.1 mL and a higher SEC retention time (indicating a smaller hydrodynamic volume). This is attributed to remaining PiBoA 2homopolymer. Integration of these regions provided quantitative information pertaining to the composition of the crude block copolymer mixture. In doing so, the remaining PiBoA in the mixture was determined to be 3.0 ± 1.0 wt%. Based upon the functionalities of the starting homopolymers (Table 1) and assuming a 100% efficient conjugation reaction, it is possible to calculate the expected composition of the crude block copolymer mixture (an example calculation is included in the ESI†). Therefore, the quantitative conjugation of PiBoA 2 and PMMA 1 would yield a mixture which would have an expected composition of 95.8 ± 0.4 wt% block copolymer, 3.0 ± 0.15 wt% PiBoA and 1.2 ± 0.24 wt% PMMA. Thus, the excellent agreement between the predicted block composition and that which was determined via the 2D chromatographic analysis confirms that the conjugation reaction (within experimental error) is quantitative, and that the remaining unreacted PiBoA homopolymer is solely due to polymer which did not bear the required dithioester functionality. Adjacent to the contour plot in Fig. 2a is a three-dimensional representation of the PMMA-b-PiBoA 5 in comparison to that of the starting PiBoA 2, which provides a very clear visualization of the efficient chemical transformation that has taken place.
 | ||
| Fig. 2 2D LACCC-SEC chromatograms and corresponding three dimensional renderings of (a) PMMA-b-PiBoA 5, (b) PMMA-b-PiBoA 6 and (c) PMMA-b-PiBoA 7; all overlayed with their respective PiBoA precursors. | ||
Moving to a higher molecular weight PiBoA, it becomes noticeable that the separation that is achieved in the SEC dimension is not as pronounced as that which can be seen in the conventional SEC traces. This can readily be explained by the fact that the 2D-chromatographic analysis of these polymers made use of a high speed SEC column (operated under a necessarily fast eluent flow rate of 3.0 mL min−1) for the second dimension. Nevertheless, a slight shift in the SEC dimension is still discernible (Fig. 2b). More importantly, the feature to take note of is the comparatively large shift in elution volume in the LACCC dimension (PiBoA 3—3.2 mL and PMMA-b-PiBoA 6—2.6 mL). The use of the high resolution SEC system (as described in the ESI†) convincingly shows the appropriate shifts in retention time.
Upon inspection of Fig. 2b, there is some overlap between the contour attributable to the block copolymer and that which is attributable to residual PiBoA. It is noted that the peaks associated with the block copolymers 6 and 7 in the LACCC analysis (Fig. 1) are slightly broader than that of 5, which contributes to this observation. However, with the aid of the superimposed contour of the precursor PiBoA 3, one can distinguish the two contours with relative ease and thus establish integration limits. It is this overlap, however, that reduces the accuracy of the integrations that may be performed on these contours. Nevertheless, integration reveals that the crude block copolymer mixture contains around 4.4 ± 1.6 wt% unreacted PiBoA 3, which is still in good agreement with the predicted value of 5.0 ± 0.25 wt%, which was determined as outlined previously.
Fig. 2c presents the 2D-chromatograms and three-dimensional renderings of the PMMA-b-PiBoA 7block copolymer and the precursor PiBoA 4. Two contours were detected in the analysis of the 2D-chromatogram of the block copolymer. The major contour centred at a LACCC elution volume of around 2.55 mL is associated with the targeted block copolymer. The minor contour centred about a LACCC elution volume of 3.1 mL is representative of remaining unreacted PiBoA 4, the amount of which was determined to be 5.2 ± 1.9 wt% by integration, which is in good agreement with the predicted value of 5.5 ± 0.83 wt% (Table 3).
| 2D LACCC-SECa/wt% | SEC deconvolutionb/wt% | Prediction based on 100% coupling efficiencyc/wt% | ||||||
|---|---|---|---|---|---|---|---|---|
| Block copolymer + PMMA | PiBoA | Block copolymer | PiBoA | PMMA | Block copolymer | PiBoA | PMMA | |
| a Determined via integration of contours in the 2D LACCC-SEC chromatograms. b Determined via integration of the deconvoluted peaks of the SEC analysis. As the iterative nature of the deconvolution minimizes error, the 1% error associated with the fit is introduced on top of the 10% error of the original SEC measurements. c Determined via calculation, taking into account the different functionalities of the respective precursors. Refer to the ESI1 for an example calculation. | ||||||||
| 5 | 97 ± 1.0 | 3.0 ± 1.0 | 95.0 | 3.5 | 1.5 | 95.8 ± 0.4 | 3.0 ± 0.15 | 1.2 ± 0.24 |
| 6 | 95.6 ± 1.6 | 4.4 ± 1.6 | 95.0 | 3.9 | 1.1 | 94 ± 0.5 | 5.0 ± 0.25 | 1.0 ± 0.2 |
| 7 | 94.8 ± 1.9 | 5.2 ± 1.9 | 94.3 | 6.2 | 0.4 | 94.2 ± 1.5 | 5.5 ± 0.83 | 0.3 ± 0.06 |
Where block copolymers are separated from their PiBoA precursors primarily in the LACCC dimension, separation of the said blocks from their PMMA precursors occurs in the SEC dimension. Fig. 3 shows the 2D-chromatograms of the synthesized block copolymers overlayed with that of the common precursor PMMA 1.‡ It can readily be observed that as the molecular weight of the block copolymer increases, their separation from PMMA 1 in the SEC dimension markedly increases. It is at this point that it should be noted that in all cases the PiBoA component of the block copolymers ranges from being roughly 1.5 times to 10 times larger than the PMMA component in terms of molecular weight. Thus, in achieving a stoichiometric ratio between the two components for the conjugation reactions, a higher mass of the PiBoA component is required than that of the PMMA component. For this reason, it is anticipated that the mass fraction of residual PMMA in the crude block copolymer mixtures will be rather small. Therefore, under the assumption of quantitative conjugation efficiency, the predicted residual PMMA compositions of the three block copolymers (PMMA-b-PiBoA 5, 6 and 7) are only 1.2 ± 0.24 wt%, 1.0 ± 0.2 wt% and 0.3 ± 0.06 wt% respectively. In the case of blocks 5 and 6, the degree of overlap between the contours of the block copolymers and the PMMA residue renders its quantification via the presently used 2D chromatographic technique not possible (Fig. 3a and b). In the case of block 7, the much larger separation as observed in Fig. 3c would be highly conducive to a quantitative analysis by integration; however, the predicted value of 0.3 ± 0.06 wt% residual PMMA was found to lie below to detection limits of the 2D LACCC-SEC system utilized. Thus the lack of any detectable contour at a LACCC elution volume of 2.5 mL and SEC retention time of 11.5 min provides a good indication that if any unreacted PMMA precursor remained in the crude reaction mixture, its quantity would be very low indeed, which supports the assumption of quantitative conjugation efficiency.
 | ||
| Fig. 3 2D LACCC-SEC chromatograms and corresponding three dimensional renderings of (a) PMMA-b-PiBoA 5, (b) PMMA-b-PiBoA 6 and (c) PMMA-b-PiBoA 7; all overlayed with their respective PMMA precursors. Note the different scales of the SEC dimension. | ||
It should be noted that the error associated with integrating such 2D-chromatograms is largely omitted from the literature. Indeed, establishing systematic error limits for such a coupled technique is no trivial task. What one can readily determine, however, is the reproducibility of the chromatographic technique. As such, after multiple analyses [eluting each sample twice and repeated re-integration (5 times) of the 2D chromatograms], the above presented compositions of the block copolymer mixtures are averages, with the maximum and minimum values providing an estimate for the (statistical) error of the analysis. As such, the (statistical) error associated with the determination of the residual PiBoA content in the three block copolymer mixtures averaged to ±35%.
While the 2D LACCC-SEC analysis of the block copolymers formed via a modular pathway proves the high efficiency of the synthesis, the analytical technique is rather labour intensive and must be fine-tuned for every individual sample that is analysed. Thus, it cannot be considered to be a standard nor a practical high throughput analytical method. In lieu of access to such a technique, it was discovered that a simple deconvolution of the conventional SEC traces of the block copolymers yielded comparably accurate composition data. Deconvolution of SEC traces has been used in the literature for the analysis of the modular construction of block copolymers and star-shaped polymers,17,38,53–58 however, its reliability has not been validated. Furthermore, in some instances in the literature, the area fractions of the deconvoluted peaks have been interpreted to give the fraction of polymer chains or the molar fraction of a certain component. This is incorrect as the SEC traces are mass distributions,§ not number distributions. As such, this technique provides mass fraction data and to interpret it in any other way is misleading and false.
In the case of block copolymer formation, the SEC traces of the precursor homopolymers are used to guide the fitting of associated peaks in the SEC trace of the crude block copolymer mixture. Fig. 4 shows the results of such a deconvolution, in which the areas under the fitted traces relate to the mass fractions of their respective components. In all cases, greater than 90 wt% of the crude block copolymer mixtures are the targeted block copolymers. Naturally, there is a limit to the sensitivity, and thus the accuracy of SEC RI traces for constituents present in minute quantities. It is therefore understandable that a deconvolution of SEC traces as presently described will likewise contain error. However, the fitting of peaks is an iterative procedure in which the error (of fitting) is reduced to a minimum, as indicated by the correlation coefficients (R2), as given in Fig. 4. In all cases, the convergence status of all fits was given as converged and all fitted data lied within 1% of the original data (as established by a 95% confidence interval). This error comes on top of the inherent error of the SEC measurement which, on the system utilized, is conservatively estimated to be 10%. It is also important to note that the deconvolution procedure is not dependent upon the SEC calibration as it is the SEC elugrams that are deconvoluted, not the calibrated molecular weight distributions.
 | ||
| Fig. 4 SEC trace deconvolution produces reliable estimations of the composition of crude block copolymer mixtures. The correlation coefficients (R2) of the peak fittings are (a) 0.998; (b) 0.999; and (c) 0.999, with 1.0 representing a perfect fit. The error in the above presented values is conservatively estimated to contain a 1% error resulting from the deconvolution on top of the 10% error of the SEC measurements (see main text for an explanation). | ||
A comparison of the composition data obtained from the 2D LACCC-SEC analysis, SEC trace deconvolution and the predicted values is presented in Table 3. Upon inspection, it is very clear that there is good agreement between the predicted values and those obtained from the two independent analytical methods. It is possible that the apparent close agreement of the presented values is pure coincidence as there is a degree of uncertainty in their determination, attributable to the inherent error of the analytical techniques and potential detector non-linearities. A seemingly attractive way around this dilemma appears to be a ‘spiking’ experiment, where known amounts of additional homopolymer are added to the analyte. However, such a procedure will not eliminate the potential disparity in (ELS) detector signal between the block copolymer and its precursors. As a 100% pure block copolymer is neither theoretically nor practically achievable for the system studied, a ‘spiking’ approach will not provide individual confirmed compositions, leaving the obtained results open to interpretation. To the best of our knowledge, the only technique potentially capable of affording unambiguous compositions with regard to the detector response is a coupling of LACCC-SEC to (quantitative) 1H-NMR spectroscopy, which allows the integration of every LACCC fraction viaNMR.59 Clearly, such an extremely complicated and expensive technique is not applicable for routine characterization efforts of block copolymers. Overall, the reproducibility within (statistical) experimental error that is observed between the three datasets reported herein suggests that there is a good correlation. It is unlikely that the deconvolution on three separate, independently prepared and analysed block copolymers samples yields individual component contributions that are by mere coincidence in agreement with the LACCC-SEC and the predicted compositions. It should be stressed that block copolymers represent some of the simplest macromolecular architectures that may be analysed by 2D LACCC-SEC. However, for much more complicated polymeric mixtures, the presently described deconvolution method would not be able to be used in its place. Nevertheless, considering the wide applicability and, therefore, importance of high purity block copolymers, the low-cost and convenient SEC deconvolution technique provides a reliable means of assessing the purity of such structures.
Conclusions
The present contribution has, for the first time, provided a chromatographic analysis of the rapid, modular construction of block copolymers, which has fast become the method of choice in the synthesis of such materials. A combination of conventional SEC, LACCC and 2D LACCC-SEC was used to quantitatively visualize the formation of novel PMMA-b-PiBoA copolymers from highly functional precursors. In addition to further confirming the quantitative nature of the conjugation chemistry utilized, characterization of the composition of the crude block copolymer mixtures was also achieved. Through this analysis and calculable predictions, the low cost deconvolution of SEC traces was found to obtain comparable composition data within experimental error.Acknowledgements
C.B.-K. acknowledges funding from the Karlsruhe Institute of Technology (KIT) within the context of the Excellence Initiative for leading German universities, the German Research Council (DFG) and the Ministry for Science and Arts of the state of Baden-Württemburg. The authors thank Mathias Glaßner for the synthesis of some of the PiBoA precursors. The authors also thank Jana Falkenhagen and Christina Schmid for helpful discussions concerning the operation and evaluation of the 2D LACCC-SEC, specifically relating to the point of the interpretation and quantification of ELSD signals.References
- V. Abetz, A. Boschetti-de-Fierro and J.-F. Gohy, in Controlled and Living Polymerizations: Methods and Applications, ed. A. H. E. Müller and K. Matyjaszewski, Wiley-VCH Verlag GmbH & Co., Weinheim, 2009 Search PubMed.
- C. B. Tang, E. M. Lennon, G. H. Fredrickson, E. J. Kramer and C. J. Hawker, Science, 2008, 322, 429–432 CrossRef CAS.
- J. Bang, U. Jeong, D. Y. Ryu, T. P. Russell and C. J. Hawker, Adv. Mater., 2009, 21, 4769–4792 CrossRef CAS.
- S. X. Ji, C. C. Liu, G. L. Liu and P. F. Nealey, ACS Nano, 2010, 4, 599–609 CrossRef CAS.
- C. Paquet and E. Kumacheva, Mater. Today, 2008, 11, 48–56 CrossRef CAS.
- K. Kataoka, A. Harada and Y. Nagasaki, Adv. Drug Delivery Rev., 2001, 47, 113–131 CrossRef CAS.
- W. A. Braunecker and K. Matyjaszewski, Prog. Polym. Sci., 2007, 32, 93–146 CrossRef CAS.
- M. Ouchi, T. Terashima and M. Sawamoto, Chem. Rev., 2009, 109, 4963–5050 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Polymer, 2008, 49, 1079–1131 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2009, 62, 1402–1472 CrossRef CAS.
- A. Okamoto, Y. Shimanuki and I. Mita, Eur. Polym. J., 1982, 18, 545–548 CrossRef CAS.
- N. Hadjichristidis, S. Pispas and G. A. Floudas, Block Copolymers: Synthetic Strategies, Physical Properties and Applications, John Wiley & Sons, Inc., Hoboken, 2002 Search PubMed.
- I. Gitsov, A. Simonyan and N. G. Vladimirov, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 5136–5148 CrossRef CAS.
- J. A. Opsteen and J. C. M. van Hest, Chem. Commun., 2005, 57–59 RSC.
- H. Durmaz, B. Colakoclu, U. Tunca and G. Hizal, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 1667–1675 CrossRef CAS.
- D. Quemener, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, Chem. Commun., 2006, 5051–5053 RSC.
- A. J. Inglis, S. Sinnwell, M. H. Stenzel and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2009, 48, 2411–2414 CAS.
- A. J. Inglis, M. H. Stenzel and C. Barner-Kowollik, Macromol. Rapid Commun., 2009, 30, 1792–1798 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- B. G. Belenky, E. S. Gankina, M. B. Tennikov and L. Z. Vilenchik, J. Chromatogr., A, 1978, 147, 99–110 CrossRef.
- G. Glockner, M. Stickler and W. Wunderlich, Fresenius' Z. Anal. Chem., 1988, 330, 46–49 CrossRef.
- G. Glockner and J. H. M. Vandenberg, J. Chromatogr., A, 1991, 550, 629–638 CrossRef CAS.
- H. Pasch, Y. Gallot and B. Trathnigg, Polymer, 1993, 34, 4986–4989 CrossRef CAS.
- P. Kilz, R. P. Kruger, H. Much and G. Schulz, Chromatographic Characterization of Polymers, Advances in Chemistry, ed. T. Provder, H. G. Barth and M. W. Urban, American Chemical Society, Washington DC, 1995, pp. 223–241 Search PubMed.
- H. Pasch, Adv. Polym. Sci., 1997, 128, 1–45 CAS.
- D. Berek, Prog. Polym. Sci., 2000, 25, 873–908 CrossRef CAS.
- D. R. Stoll, X. P. Li, X. O. Wang, P. W. Carr, S. E. G. Porter and S. C. Rutan, J. Chromatogr., A, 2007, 1168, 3–43 CrossRef CAS.
- P. Dugo, F. Cacciola, T. Kumm, G. Dugo and L. Mondello, J. Chromatogr., A, 2008, 1184, 353–368 CrossRef CAS.
- D. Berek, Anal. Bioanal. Chem., 2010, 396, 421–441 CrossRef CAS.
- W. Lee, D. Y. Cho, T. Y. Chang, K. J. Hanley and T. P. Lodge, Macromolecules, 2001, 34, 2353–2358 CrossRef CAS.
- I. Park, S. Park, D. Y. Cho, T. Y. Chang, E. Kim, K. Y. Lee and Y. J. Kim, Macromolecules, 2003, 36, 8539–8543 CrossRef CAS.
- H. J. A. Philipsen, J. Chromatogr., A, 2004, 1037, 329–350 CrossRef.
- M. Li, N. M. Jahed, K. Min and K. Matyjaszewski, Macromolecules, 2004, 37, 2434–2441 CrossRef CAS.
- K. Min, H. F. Gao and K. Matyjaszewski, J. Am. Chem. Soc., 2005, 127, 3825–3830 CrossRef CAS.
- H. F. Gao, K. Min and K. Matyjaszewski, Macromol. Chem. Phys., 2006, 207, 1709–1717 CrossRef CAS.
- J. Falkenhagen, H. Much, W. Stauf and A. H. E. Müller, Macromolecules, 2000, 33, 3687–3693 CrossRef CAS.
- V. Mass, V. Bellas and H. Pasch, Macromol. Chem. Phys., 2008, 209, 2026–2039 CrossRef CAS.
- A. J. Inglis, S. Sinnwell, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, Macromolecules, 2008, 41, 4120–4126 CrossRef CAS.
- L. Nebhani, S. Sinnwell, A. J. Inglis, M. H. Stenzel, C. Barner-Kowollik and L. Barner, Macromol. Rapid Commun., 2008, 29, 1431–1437 CrossRef CAS.
- S. Sinnwell, A. J. Inglis, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, Chem. Commun., 2008, 2052–2054 RSC.
- S. Sinnwell, A. J. Inglis, M. H. Stenzel and C. Barner-Kowollik, Macromol. Rapid Commun., 2008, 29, 1090–1096 CrossRef CAS.
- L. Nebhani, S. Sinnwell, C. Y. Lin, M. L. Coote, M. H. Stenzel and C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 6053–6071 CrossRef CAS.
- T. Paulöhrl, A. J. Inglis and C. Barner-Kowollik, Adv. Mater., 2010, 22, 2788–2791 CrossRef.
- I. Abrunhosa, M. Gulea and S. Masson, Synthesis, 2004, 6, 928–934.
- A. Alberti, M. Benaglia, M. Guerra, M. Gulea, P. Hapiot, M. Laus, D. Macciantelli, S. Masson, A. Postma and K. Sparnacci, Macromolecules, 2005, 38, 7610–7618 CrossRef CAS.
- C. Schmid, J. Falkenhagen and C. Barner-Kowollik, J. Polym. Sci. –Polym. Chem., 2010 Search PubMed , submitted.
- E. H. H. Wong, M. H. Stenzel, T. Junkers and C. Barner-Kowollik, Macromolecules, 2010, 43, 3785–3793 CrossRef CAS.
- W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2008, 29, 952–981 CrossRef CAS.
- C. Barner-Kowollik, Macromol. Rapid Commun., 2009, 30, 1625–1631 CrossRef CAS.
- A. J. Inglis, T. Paulohrl and C. Barner-Kowollik, Macromolecules, 2010, 43, 33–36 CrossRef CAS.
- S. P. S. Koo, T. Junkers and C. Barner-Kowollik, Macromolecules, 2009, 42, 62–69 CrossRef CAS.
- F. Günzler, E. H. H. Wong, S. P. S. Koo, T. Junkers and C. Barner-Kowollik, Macromolecules, 2009, 42, 1488–1493 CrossRef CAS.
- O. Altintas, B. Yankul, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 6458–6465 CrossRef CAS.
- H. F. Gao and K. Matyjaszewski, Macromolecules, 2006, 39, 4960–4965 CrossRef CAS.
- H. Durmaz, A. Dag, O. Altintas, T. Erdogan, G. Hizal and U. Tunca, Macromolecules, 2007, 40, 191–198 CrossRef CAS.
- O. Altintas, A. L. Demirel, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5916–5928 CrossRef CAS.
- O. Altintas, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 1218–1228 CrossRef CAS.
- H. Durmaz, A. Dag, A. Hizal, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7091–7100 CrossRef CAS.
- W. Hiller, H. Pasch, P. Sinha, T. Wagner, J. Thiel, M. Wagner and K. Müllen, Macromolecules, 2010, 43, 4853–4863 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Add experimental details, 1H-NMR and ESI-MS spectra of the synthesized homopolymers. See DOI: 10.1039/c0py00189a |
| ‡ Ideally, Fig. 2 and 3 should be combined, however, the PSS software used to process the presented data does not allow for the overlay of more than one 2D-chromatogram. |
| § More specifically, SEC traces from RI-detection are relative to both mass and incremental changes in refractive index. |
| This journal is © The Royal Society of Chemistry 2011 |