Scope and limitation of the copper free thermal Huisgen cross-linking reaction to stabilize the chromophores orientation in electro-optic polymers
Annabelle
Scarpaci†
a,
Clément
Cabanetos†
a,
Errol
Blart
a,
Yann
Pellegrin
a,
Véronique
Montembault
b,
Laurent
Fontaine
*b,
Vincent
Rodriguez
*c and
Fabrice
Odobel
*a
aUniversité de Nantes, CNRS, Chimie et Interdisciplinarité: Synthèse, Analyse, Modélisation (CEISAM), UMR CNRS n° 6230, 2, rue de la Houssinière—BP 92208, 44322, Nantes Cedex 3, France. E-mail: Fabrice.Odobel@univ-nantes.fr; Fax: +33 (0)2 51 12 54 02; Tel: +33 (0)2 51 12 54 29
bLCOM-Chimie des Polymères, UCO2M, Université du Maine, UMR CNRS 6011, Avenue O. Messiaen, 72085, Le Mans Cedex 9, France. E-mail: laurent.fontaine@univ-lemans.fr; Fax: +33 (0)2 43 83 37 54; Tel: +33 (0)2 43 83 33 25
cUniversité de Bordeaux, Institut des Sciences Moléculaires, UMR5255 CNRS, 351 cours de la Libération, 33405, Talence Cedex, France. E-mail: v.rodriguez@ism.u-bordeaux1.fr
First published on 13th October 2010
Abstract
New methacrylate copolymers incorporating two complementary thermally cross-linkable groups (azide or ethynyl) for implementation in electro-optic devices were synthesized and their nonlinear optical properties were investigated. These copolymers were prepared from a monomer containing Disperse Red 1 (DR1) as active NLO chromophore which is end substituted either by an azide or ethynyl group connected via a rigid (phenyl) or flexible spacer (alkyl chain). The second monomer is either a trimethylsilyl-propargyl methacrylate, or an azidopropyl methacrylate or a trimethylsilyl-phenyl methacrylate. The determination of the reactivity ratios showed that the monomer containing the DR1 chromophore is more reactive than trimethylsilyl-propargyl methacrylate. The cross-linking temperatures of these polymers range from 150 °C to 187 °C depending on the rigidity of the spacers connecting the cross-linkable units. These polymers displayed relatively high macroscopic electro-optic stability, enhanced upon cross-linking by more than 40 °C relative to non-cross-linked polymers. The results underscore the importance of the flexibility of the spacers to achieve the stable bulk electro-optic response. While rigidity is favorable to maintain the orientation of the chromophores, the optimal polymer is the one containing a flexible and a rigid spacer, since the mobility of the reactive groups is a key parameter which guarantees a high cross-linking conversion within the polymer. This study demonstrates the versatility of this new cross-linking process because we showed that the reactive groups (azide or trimethylsilylacetylated groups) can be interconverted (on the chromophore or as polymer side chain) with no change on the overall electro-optic activity and its thermal stability. Furthermore, preliminary kinetic study indicates that the Huisgen reaction rate can be controlled by the substituent on the ethynyl group opening the possibility to tune the cross-linking temperature by the careful choice of this substituent.
Introduction
Remarkable progress has been made in the past decades to optimize the properties of nonlinear optical (NLO) polymers since they represent promising low cost materials for the fabrication of high-speed electro-optic (EO) devices such as Mach–Zehnder modulators.1–4 The key advantages of EO organic polymers over the commonly used lithium niobate (LiNbO3) reside in their very large EO coefficients (up to 300 pm V−1), high bandwidth capacity (greater than 10 GHz), large processability and low dielectric constant.1 However, one of the most challenging issues limiting the utilization of organic materials for practical applications is the relatively low temporal and thermal stability of the non-centrosymmetric alignment of the chromophores.5 Indeed, after poling, the chromophores tend to relax and aggregate by intermolecular electrostatic interactions owing to their dipolar nature. As a result, this phenomenon leads to the loss of the macroscopic NLO activity. To solve this problem, two main strategies have been undertaken. Firstly, using high glass transition temperature (Tg) polymers such as polyimides or polyurethanes, especially when the NLO chromophores are chemically bonded to the polymer backbones, which enables to design materials exhibiting a quite stable electro-optic activity.3,6 However, the high temperatures of the polymer synthesis or the poling process can induce the decomposition or the sublimation of the chromophores. The second strategy consists in bringing about a cross-linking reaction within the polymer matrix to harden the material and to freeze the chromophores orientation after the poling process. Among the most used cross-linking reactions, the anthracene–maleimide Diels–Alder cyclo-addition,1,7 the cyclodimerization of trifluorovinylether,8 the thermal decomposition of cyclobutanone9 and the epoxide opening with a carboxylic acid10,11 were implemented to electro-optic polymers resulting in a very significant enhancement of the stability of the macroscopic NLO activity. In a recent communication, we reported the utilization of the copper-free Huisgen thermal 1,3-dipolar cycloaddition reaction as an attractive cross-linking system for EO materials.12 The synthesis of the methacrylate type polymer P1, resulting from the copolymerization of an azide derivatized DR1 chromophore and an acetylated monomer, was performed (Fig. 1).12 The copper-catalyzed Huisgen 1,3-dipolar cycloaddition, referred as “click reaction”, is very well-known, as epitomized by the large number of recent reports in this area. It has been intensively employed to functionalize polymers13 or surfaces14 with various molecular units or simply to synthesize new triazole derivatives for pharmaceutical applications.15 Mixtures of polyalkyne and polyazide derivatives were employed as adhesive glues to stick together two copper surfaces.16 Recently, site isolation groups were grafted on NLO chromophores,17 and precursor polymers were post-functionalized using the same approach.18,19 The thermal version/pathway of the 1,3-dipolar cycloaddition was reported by Eastman Kodak researchers in the early eighties to post-functionalize poly(vinyl-benzyl azide) with various acetylenic derivatives.20 More recently, this cycloaddition reaction has been extended by using copper-free click ligation of azide substrates to strained alkynes making it applicable to biological environments for which copper salt could be noxious.21 The absence of any metal-based additive to catalyze the reaction is a requirement for the applications in EO, because these substances would certainly enhance the conductivity of the polymer films and would induce dielectric breakdowns upon poling during the application of a high voltage. Our first results have shown that copper-free thermal Huisgen reaction proved to be particularly efficient to stabilize the chromophore orientation after the poling step since the EO properties of the polymer P1 are maintained up to 137 °C, which suits the generally accepted requirements for practical applications.12 | ||
| Fig. 1 Structures of the polymers described in this study. | ||
In this present paper, we studied the versatility and the scope of this cross-linking system. More precisely, the impact of two structural modifications on the poling efficiency and on the overall thermal stability of the chromophores orientation was investigated. Firstly, we inverted the position of the azide and the alkyne groups, which were initially borne by the chromophore and by a side chain of the polymer backbone, respectively (P1versusP2 in Fig. 1). Secondly, we explored the influence of the rigidity of the spacer between the reactive cross-linking groups (azido or ethynyl) by using either an alkyl or an aryl connector. To this end, starting with fully flexible spacers (P2 and P3), we rigidified the spacer either only on the chromophore (P1) or on the chromophore and on the side chain (P4). The results of this study demonstrate the versatility of this new cross-linking process which underscores the importance of the connectors' flexibility to obtain a stable bulk electro-optic response. The optimal system is the one containing one flexible and one rigid spacer. This experimental fact is rationalized if we consider that the mobility of the reactive groups is a key parameter to warrant a high cross-linking efficiency within the polymer.
Experimental part
Materials
4-N-Ethyl-N-(2-azidoethyl)amino-4′-nitroazobenzene 6,12chromophore 10,123-azidopropanol, 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methyl-morpholinium chloride (DMTMM),222-methacrylic acid 3-trimethylsilanyl-prop-2-ynyl ester 13,193-azidopropyl methacrylate 1423 and 4-trimethylsilethynyl-phenol 1524 were prepared according to the methods described in the literature. Azobisisobutyronitrile (AIBN) initiator was purified by recrystallization in methanol before use. Methylmethacrylate acid (MMA) was distilled under reduced pressure before use to remove the inhibitor. Trimethylsilyl propargyl alcohol was purchased from Aldrich. All other chemicals were purchased from Acros and used as received.Methods
The NMR spectra were recorded using a Bruker 300 MHz instrument (ARX-300, Bruker) with tetramethylsilane (TMS) as the internal standard and CDCl3 as the solvent. The IR spectra were recorded using a FTIR spectrometer (Clark-MXR CPA). UV-Visible absorption spectra were recorded on a UV-2401PC Shimadzu spectrophotometer. Thermal analyses were performed using a TA instruments Q100 in a nitrogen atmosphere at a heating rate of 10 °C min−1. The number- and weight-averaged molecular weights and molecular weight distributions were determined using a size exclusion chromatography (SEC) on a system equipped with a SpectraSYSTEM AS 1000 autosampler, with a guard column (Polymer Laboratories, PL gel 5 µm guard column) followed by two columns (Polymer Laboratories (PL), 2 PL gel 5 µm MIXED-D columns), with a SpectraSYSTEM RI-150 detector. Tetrahydrofuran (THF) was used as the mobile phase at a flow rate of 1 mL min−1 at 35 °C. Polystyrene standards (580–483 × 103 g mol−1) were used to calibrate the SEC. High resolution electro-spray mass spectra (HR-ESMS) were collected in positive mode on a MS/MS ZABSpec TOF of Micromass equipped with a geometry EBE TOF. The samples were injected in dichloromethane. Elemental analyses were carried out by combustion using a CHN 2400 analyzer for carbon, hydrogen and nitrogen and by pyrolysis using an O Vario EL III analyzer for oxygen. Column chromatography was carried out either with Merck 5735 Kieselgel 60F (0.040–0.063 mm mesh). Air sensitive reactions were carried out under argon in dry solvents.NLO measurements
HRS experiments were performed on dilute solutions (between 10−4 and 10−6 M) in acetonitrile, which correspond to typical concentrations used to check the linear dependency of absorbance and determine the extinction coefficient ε. In this NLO scattering technique, the intensity of the incoherent scattered light at the second harmonic frequency of an IR Nd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :YAG pulsed laser (picosecond regime) is used to determine the first hyperpolarizability β. The scattered harmonic light is related to quadratic products between components of the molecular β-tensor that correspond to isotropic averaging over the molecular motions (non-interacting molecules). Assuming a pseudo C2v molecular symmetry where the molecule lies in a mean (xz) plane (with z the two-fold symmetry axis), and assuming Kleinman symmetry, two independent components βzxx and βzzz remain.25 These components are determined through polarization scan measurements that involve linear, circular and general elliptical state of polarization of the incident beam. More details about the procedure can be found in ref. 26. For every chromophore, the βzzz components were at least one order of magnitude larger than the βxzz component. These results warrant the validity of our previous assumption of the so-called Kleinman condition that states that βzxx = βxxz since off-diagonal components are indeed weak with respect to the longitudinal one which is actually enhanced by resonance effects.
:YAG pulsed laser (picosecond regime) is used to determine the first hyperpolarizability β. The scattered harmonic light is related to quadratic products between components of the molecular β-tensor that correspond to isotropic averaging over the molecular motions (non-interacting molecules). Assuming a pseudo C2v molecular symmetry where the molecule lies in a mean (xz) plane (with z the two-fold symmetry axis), and assuming Kleinman symmetry, two independent components βzxx and βzzz remain.25 These components are determined through polarization scan measurements that involve linear, circular and general elliptical state of polarization of the incident beam. More details about the procedure can be found in ref. 26. For every chromophore, the βzzz components were at least one order of magnitude larger than the βxzz component. These results warrant the validity of our previous assumption of the so-called Kleinman condition that states that βzxx = βxxz since off-diagonal components are indeed weak with respect to the longitudinal one which is actually enhanced by resonance effects.
SHG measurements were performed using the optical setup described in a previous study.27 Polarized SHG Maker fringe patterns were recorded after the poling process, using a 1064 nm Nd:YAG laser operating at very low irradiance (pulse energy < 20 µJ; repetition rate 50 Hz; pulse width 15 ns). A general SHG transfer matrix has been applied, allowing experimental determination of the resonance-enhanced NLO coefficients dij, as well as the linear absorption coefficients of the harmonic wave (532 nm) in the parallel (α∥) and (α⊥) direction with respect to the poling field. Thus, anisotropic absorption of the harmonic wave was explicitly taken into account and SHG coefficients were determined using the quartz reference with coefficient d11 = 0.3 pm V−1 at 1064 nm. More details about the general procedure to determine the linear and nonlinear optical constants can be found elsewhere.28
Synthesis of the materials
:1 by volume, 62.4 mL) was added sodium hydride (60% suspension in mineral oil, 552.8 mg, 13.82 mmol, 1.3 equiv.) at 0 °C, and the mixture was stirred at this temperature for 30 min. Benzyl bromide was then added dropwise (1.39 mL, 1.1 equiv.) at 0 °C, and the resulting mixture was stirred from 0 °C to room temperature for one night. The reaction was quenched by the addition of aqueous NH4Cl and extracted several times with diethyl ether. The combined organic phases were dried on MgSO4, filtered and evaporated to give the crude product, which was purified by silica gel column chromatography using dichloromethane/petroleum ether (20/80) as eluting system. Compound 2a was obtained as a yellow oil (1.355 g, 87%). 1H NMR (300 MHz, CDCl3), δ (ppm): 7.26–7.35 (m, 5H); 4.64 (s, 2H); 4.20 (d, 2H, J = 2.5 Hz); 2.5 (t, 1H, J = 2.5 Hz). 13C NMR (75 MHz, CDCl3), δ (ppm): 137.4; 128.5; 128.2; 128; 79.7; 74.7; 71.6; 57.1. EI-MS: m/z calcd for C10H10O: 146.1 (M+), found: 146.1. FT-IR (KBr, cm−1): 2966, 2851 (νst(CH2)); 2118 (νw(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C)); 1608 (νst(C
C)); 1608 (νst(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C)).
C)); 1609 (νst(CC)).
C)); 1616 (νst(CC)).
C)); 1607 (νst(CC)).
N)); 1957 (νw(CC)); 1605 (νst(CC)).
C)).
C)); 1609 (νst(CC)).
C)); 1616 (νst(CC)).
C)); 1607 (νst(CC)).
N)); 1957 (νw(CC)); 1605 (νst(CC)).
General procedure for the synthesis of compounds 3 and 4
The alkyne moiety 2 (typically 1 mmol) and 1 (1 mmol) were dissolved in 1,2-dichlorobenzene (2 mL) and heated at different temperatures. In all cases, after removal of the solvents, the crude product was purified by column chromatography on silica gel eluted with dichloromethane to give the 1,4 (3) and the 1,5 (4) isomers of the adducts as red powders.500).
400).
000).
000).
000).
000).
000).
600).
500).
Isomer 1,4 of compound 3aviacopper-catalyzed Huisgen cycloaddition
The alkyne 2a (100 mg, 0.685 mmol, 1.0 equiv.) and the chromophore 1 (233 mg, 0.685 mmol, 1.0 mmol) were suspended in a 1:1 mixture of water and tert-butyl alcohol (5 mL). Sodium ascorbate (1 M in water, 0.068 mmol, 68.5 µL) was added, followed by copper sulfate pentahydrate (1.75 mg, 6.85 × 10−3 mmol, 1 mol%). The heterogeneous mixture was stirred at r.t. overnight. After the removal of the solvents, the crude product was purified by column chromatography on silica gel eluted with dichloromethane to give the 1,4 isomer as red powder (318 mg, 96%).
1H NMR (300 MHz, CDCl3), δ (ppm): 8.33 (d, 2H, J = 8.8 Hz); 7.91 (d, 2H, J = 8.8 Hz); 7.89 (d, 2H, J = 9 Hz); 7.48 (s, 1H); 7.28–7.36 (m, 5H); 6.72 (d, 2H, J = 9.3 Hz); 4.67 (s, 2H); 4.60 (t, 2H, J = 6.6 Hz); 4.55 (s, 2H); 3.96 (t, 2H, J = 6.6 Hz); 3.31 (q, 2H, J = 6.6 Hz); 1.15 (t, 3H, 6.6 Hz). HRMS-CI: m/z calcd for C26H28N7O3: 486.2254 (MH+), found: 486.2255. UV-Vis (CH2Cl2): λmax (ε (mol−1 L cm−1)) = 463 nm (34500).
General procedure for the esterification of carboxylic acid 6 with alcohol derivatives 8 and 9
To a solution of carboxylic acid compound 6 (1 equiv., typically 750 mg) and alcohol 8 or 9 (1.2 equiv.) in 4 mL of THF was added N-methylmorpholine (3 equiv.) and then dimethoxy triazine-N-methyl morpholinium chloride (1.1 equiv.). The solution was stirred for one night at room temperature. Solvents were then evaporated under reduced pressure to give a crude product, which was purified by silica gel column chromatography using dichloromethane/petroleum ether (8/2) as eluent leading to compounds 11 and 12.O)); 1598 (νst(CC)). UV-Vis (CH2Cl2): λmax (ε (mol−1 L cm−1)) = 480 (29600).
C)); 1726 (νst(CO)); 1598 (νst(CC)). UV-Vis (CH2Cl2): λmax (ε (mol−1 L cm−1)) = 482 (31600).
1H NMR (300 MHz, CDCl3), δ (ppm): 7.49 (m, 2H); 7.08 (m, 2H); 6.34 (s, 1H); 5.76 (s, 1H); 2.54 (s, 3H); 0.25 (s, 9H). 13C NMR (75 MHz, CDCl3), δ (ppm): 163.02; 150.73; 135.68; 133.14; 127.51; 121.57; 121.55; 104.24; 53.41; 17.38; 3.51. EI-MS: m/z calcd for C15H18O2Si: 258.10 (MH+), found: 259.19. FTIR (KBr, cm−1): 2150 s (CC).
General procedure for the polymerization of the cross-linkable polymers
Compound 10, 11 or 12 (3 equiv., generally 600 mg), methacrylate monomer 13, 14 or 16 (7 equiv.) and AIBN (8% by weight from the total amount of monomers) were dissolved in 8 mL of tetrahydrofuran in a dry Schlenk tube. The mixture was degassed by three freeze–pump–thaw cycles, and heated for 18 hours at 70 °C in the dark. Once back to room temperature, the mixture was poured dropwise in a large amount of methanol (80 mL), yielding a red precipitate. The solid was washed twice and isolated by centrifugation. It was then dried for 12 hours under reduced pressure at 35 °C. A red powder was obtained.C)); 2098 (νst(N3)); 1732 (νst(CO)); 1598 (νst(CC)). UV-Vis (CH2Cl2): λmax (%w) = 476 nm (42). TGA-DSC (10 °C min−1): Tg = 89 °C; Tc = 168 °C; Td = 263 °C. Anal. calcd for x = 3; y = 7: C, 60.10; H, 6.43; N, 11.29. Found: C, 59.97; H, 6.29; N, 11.66.
500; Ip = 1.4. FT-IR (KBr, cm−1): 2958 (νst(CH2)); 2183 (νst(CC)); 2098 (νst(N3)); 1728 (νst(CO)); 1598 (νst(CC)). UV-Vis (CH2Cl2): λmax (%w) = 482 nm (53). TGA-DSC (10 °C min−1): Tg = 87 °C; Tc = 168 °C; Td = 249 °C. Anal. calcd for x = 3; y = 7: C, 55.54; H, 6.23; N, 16.70. Found: C, 56.54; H, 6.43; N, 13.93.
C)); 2098 (νst(N3)); 1727 (νst(CO)); 1598 (νst(CC)). UV-Vis (CH2Cl2): λmax (%w) = 478 nm (43). TGA-DSC (10 °C min−1): Tg = 87 °C; Tc = 150 °C; Td = 285 °C. Anal. calcd for x = 3; y = 7: C, 59.70; H, 6.88; N, 10.30. Found: C, 58.85; H, 6.89; N, 10.36.
C)); 2098 (νst(N3)); 1727 (νst(CO)); 1598 (νst(CC)). UV-Vis (CH2Cl2): λmax (%w) = 483 nm (46). TGA-DSC (10 °C min−1): Tg = 90 °C; Tc = 187 °C; Td = 254 °C. Anal. calcd for x = 3; y = 7: C, 65.06; H, 6.07; N, 9.74. Found: C, 63.68; H, 5.91; N, 9.55.
Results and discussion
Kinetic studies on the Huisgen reaction in solution
It is well-known that the kinetic rate of the 1,3-dipolar cycloaddition depends on the substituents on the alkyne and/or the azide reagents.29 This characteristic could open the possibility to tune the crosslinking temperature (Tc) by changing the substituent on the ethynyl group in order to fit the glass temperature (Tg) of a given polymer. Indeed, Tc is a critical parameter to optimize the poling process, since the cross-linking reaction should occur close to the Tg. Indeed, a premature cross-linking (starting much below Tg) would prevent an efficient poling owing to a too early hardening of the matrix. On the other hand, cross-linking the polymer at a temperature much above the Tg usually decreases the poling efficiency owing to randomization of the chromophore orientation by thermal agitation. Accordingly, we initially undertook a kinetic study in order to determine the effects of the size and the electronic properties of the substituent on the ethynyl group on the rate of the cross-linking reaction (Scheme 1). The study of the Huisgen reaction's kinetic rates was carried out in solution using an azido functionalized DR1 derivative 1 and the alkynes 2a–e substituted by different R groups (Scheme 1). The R substituents differ by their bulkiness and their electronic properties (electron withdrawing or donating effects). The reaction was conducted in o-dichlorobenzene, a low dielectric constant solvent mimicking a methacrylate polymer matrix environment with a high boiling temperature (200 °C) enabling us to perform the kinetic study on a large range of temperatures.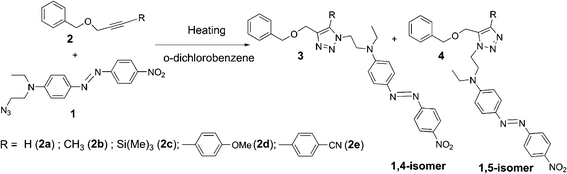 | ||
| Scheme 1 Reaction involved for the kinetic studies. | ||
The reaction was initially investigated at different temperatures using the terminal alkyne derivative (2a: R = H, Scheme 1) and the conversion rates were determined by monitoring the formation of triazole units by 1H NMR. The surfaces of the signals at 7.48 ppm and 6.61 ppm, corresponding to the hydrogen groups of the 1,4- and 1,5-isomers of the 1,2,3-triazoles 3 and 4 respectively, could be cleanly integrated, since they do not overlap with other signals. The assignment of the signal at 7.48 ppm to the 1,4 isomer was deduced from the preparation of a pure sample of the 1,4-isomer of adduct 3a catalyzed with CuSO4·5H2O in the presence of sodium ascorbate (see Experimental part). The absence of regioselectivity during the copper free thermally induced 1,3-dipolar cycloaddition was confirmed by the formation of an equimolar mixture of 1,4- and 1,5-regioisomers 3 and 4.
At 100 °C, the reaction between 1 and 2a was very slow since the yield of the detected cyclo-products after one hour was lower than 5%. Nevertheless, faster rates were observed between 160 °C and 180 °C since quasi-quantitative conversion was reached within 5 hours and 1 hour respectively (Fig. 2). In order to better distinguish the relative reactivity of the different alkynes, the temperature of 140 °C was chosen for the kinetic study with the other alkynes 2b–e (Fig. 3).
 | ||
| Fig. 2 Kinetic traces of the reaction between 1 and 2a in o-dichlorobenzene at different temperatures. | ||
 | ||
| Fig. 3 Kinetic traces of the reaction between 1 and the alkynes 2a–e substituted with different R groups. The reactions were run in o-dichlorobenzene at 140 °C. | ||
Fig. 3 shows that the most reactive alkyne in this series is the terminal alkyne 2a, but its rate is very close to the TMS substituted alkyne 2c. The high reactivity of terminal alkyne 2a probably stems from its large accessibility, while the silane activates the alkyne. It is known that 1,3-dipolar cycloaddition reaction is faster with electron withdrawing substituents on the alkyne, while electron releasing groups induce the reverse effect.29 This general view is supported by our results since the bulky and electron rich p-methoxyphenyl group (2d) gives the slowest rate among the series, while the reaction with p-cyanophenyl substituted alkyne (2e) is faster. These results indicate that the rate of the cycloaddition reaction between 1 and 2a–e is very sensitive to the steric hindrance of the R substituent on the alkyne, although the electronic properties play an important role as well. Let us note that 2a and 2c both show a similar reactivity towards the azide derivatized NLO chromophore, underlining that the deprotection step of the TMS–polymer is not necessary.23,30 In conclusion, this study demonstrates that it should be possible to tune the cross-linking rate, and thus the temperature, of the system by changing the nature of the R group on the alkyne underlying that it is certainly possible to implement this cross-linking reaction with a variety of polymer backbones presenting different Tgs.
Monomers and polymers syntheses
The methacrylate monomers 10, 11 and 12 containing the chromophore were synthesized in a nearly quantitative yield by amidification or esterification of the carboxylic acid derivative 6 which was initially activated by 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methyl morpholinium chloride (DMTMM) in the presence of N-methylmorpholine (NMM) and further reacted with 4-azidoaniline hydrochloride 7, 4-azidopropanol 8, or trimethylsilyl propargyl alcohol 9 (Scheme 2). The polymers P1–P4 were obtained in 50 to 79% yield by free-radical copolymerization initiated by AIBN at 70 °C of the above monomers with the complementary monomer 13, 14 or 16 (Schemes 3–5). | ||
| Scheme 2 Synthesis of methacrylate monomers 10–12 functionalized by the chromophore. | ||
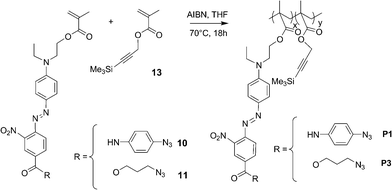 | ||
| Scheme 3 Preparation of the polymers P1 and P3. | ||
 | ||
| Scheme 4 Preparation of the polymer P2. | ||
 | ||
| Scheme 5 Preparation of the polymer P4. | ||
Polymers
P1–P3 are highly soluble in common polar organic solvents, such as CH2Cl2, CHCl3, THF, DMF and DMSO, but polymer P4 is much less soluble in these solvents. The number-average molecular weight (Mn) and the polydispersity index (PDI) referenced versuspolystyrene standards were determined by size exclusion chromatography (Table 2). The chromophore loading levels were measured by three different methods, namely: measuring the absorbance of the chromophore at 450 nm, the integration of 1H NMR signals and the elemental analyses. The results of these different analyses are consistent with one another and give a percentage of chromophore in the polymers between 43 wt% and 58 wt%. The FTIR spectra clearly revealed the presence of an intense azide absorption band around 2180 cm−1 and a weak CC absorption band around 2260 cm−1 indicating that the cross-linking reaction has not occurred during the polymerization reaction.
Determination of the monomer reactivity ratios
Polymer P1 giving the best results in terms of poling efficiency and orientation stability (see below), we therefore decided to determine the monomer reactivity ratios of 10 and 13 in order to infer their mean distribution along the polymer backbone. The placement of the chromophores relative to the propargyl units (statistic or block polymers) is certainly an important piece of information to rationalize the overall properties of the polymer, because the close packing of the chromophores should impact the bulk electro-optic activity of the polymer. To this end, different ratios of the monomers 10 and 13 were polymerized in the same conditions as that used for the preparation of the polymer, but the reaction was stopped at low conversion (below 10%) and the composition of the resulting polymer was determined by 1H NMR (Table 1). The experimental results were analyzed with the methods of Fineman-Ross31 and Kelen–Tüdös32 to calculate the monomer reactivity ratios. Both methods give the same reactivity ratio index values, which are: r1010 = 1.041 ± 0.001 and r1313 = 0.009 ± 0.001.| Feeding molar ratio of the monomers | Molar fraction in the copolymera | Conversionb (%) | ξ c | η c | ||
|---|---|---|---|---|---|---|
| 10 | 13 | DR1N3 | Propargyl | |||
| a Mean value of the composition determined by 1H NMR and by absorption spectrophotometry. b Based on the yield determined with the mass of polymer obtained after precipitation in methanol. c ξ indicates the molar conversion of monomer 10 and η the molar conversion of the monomer 13. n. d. = not determined owing to insolubility. | ||||||
| 0.1 | 0.9 | 0.54 | 0.46 | 5 | 0.029 | 0.046 |
| 0.2 | 0.8 | 0.61 | 0.39 | 4.8 | 0.103 | 0.232 |
| 0.3 | 0.7 | 0.62 | 0.38 | 8.2 | 0.244 | 0.359 |
| 0.4 | 0.6 | 0.64 | 0.36 | 7.8 | 0.417 | 0.486 |
| 0.5 | 0.5 | 0.67 | 0.33 | 8.5 | 0.585 | 0.603 |
| 0.6 | 0.4 | 0.71 | 0.29 | 9.5 | 0.725 | 0.699 |
| 0.7 | 0.3 | 0.77 | 0.23 | 8.2 | 0.823 | 0.828 |
| 0.8 | 0.2 | 0.84 | 0.16 | 7.8 | 0.897 | 0.953 |
| 0.9 | 0.1 | n.d. | n.d. | 8 | — | — |
The fact that r1010 ≈ 1 indicates that the radical generated from 10 reacts slightly faster with 10 than with 13. Conversely, r1313 being <1 the radical generated from 13 will preferably react with 10 than with 13. The steric hindrance of 13 might explain its lower reactivity with respect to 10. Taking into account the values of the reactivity ratio of 10 and 13 and feed ratio (13/10 = 7/3) for the preparation of P1 the simulation of the monomer distribution in the polymer chain can be deduced.33 Taking into account that molar concentration of the chromophores in the polymer P1 is much lower than that of propargyl methacrylate, the simulation indicates that, at maximum, about 1.4 chromophores are statistically neighbored with 1 propargyl unit.
Determination of the glass and cross-linking temperatures of the polymers
The polymers were studied by TGA to determine the decomposition temperature (Td) and by differential scanning calorimetry (DSC) to determine the glass transition temperature (Tg) and the cross-linking temperature (Tc). A typical DSC trace is showed in Fig. 4. | ||
| Fig. 4 Differential scanning calorimetry (DSC) trace of polymer P2 recorded under nitrogen flow with a scanning rate of 10 °C min−1. | ||
For P1–P4, the DSC traces feature a first breakdown around 90 °C, which is attributed to the glass transition temperatures (Tg). Then, a first exothermic peak assigned to the Huisgen cross-linking reaction appears between 150 °C and 187 °C.34 It should be pointed out that the cross-linking temperature of polymers P1 and P4 is higher than that of P2 and P3 probably because the rigid aryl spacer reduces the mobility of the reacting groups (azide and/or alkyne). Furthermore, the phenyl azide in P1 and P4, which is more electron deficient than the alkyl azide in P2 or P3, is certainly less reactive. A very intense exothermic peak between 249 °C and 285 °C is attributed to the decomposition of triazole probably into nitrene.35 A similar exothermic reaction was previously reported in triazole based polymers by Ergin and co-workers.34 However, thermogravimetric analyses showed that all the polymers exhibit high thermal stability since the decomposition temperatures (Tds) are greater than 250 °C (Table 2).
| Entry | Polymer | M n/g mol−1 | M w/g mol−1 | PDI | T g a/°C | T c b/°C | T d c/°C | d d/µm | d 33 e/pm V−1 |
|---|---|---|---|---|---|---|---|---|---|
| a T g: glass transition temperature. b T c: cross-linking temperature. c T d: decomposition temperature (Td measured at 5 wt% decomposition). d d: thickness of the film. e d 33: coefficient measured at 1064 nm. | |||||||||
| 1 | P1 | 7100 | 13500 |
1.9 | 89 | 168 | 263 | 2.7 | 50 ± 5 |
| 2 | P2 | 18500 |
25900 |
1.4 | 87 | 157 | 249 | 1.7 | 53 ± 5 |
| 3 | P3 | 9200 | 12800 |
1.4 | 87 | 150 | 285 | 0.3 | 60 ± 6 |
| 4 | P4 | 7100 | 20800 |
2.9 | 90 | 187 | 254 | 0.2 | 19 ± 3 |
Nonlinear optical measurements
Initially, the first hyperpolarizability of the chromophores 10–12 was measured by hyper-Rayleigh scattering (HRS) technique in acetonitrile at 1064 nm with a Nd:YAG laser (Table 3). The static β0 values were also estimated by using the two-state model.36 Interestingly, the first hyperpolarizabilities of the new chromophores 10–12 are substantially lower than that of DR1, which is probably due to the change of the nitro group from the para to the ortho position.
| Entry | Chromophore | β zzz (10−30 esu) at 1064 nm | β 0 (10−30 esu) | λ max/nm; ε/mol−1 L cm−1 |
|---|---|---|---|---|
| a DR1: N-Ethyl-N-(2-hydroxyethyl)-4-(4-nitrophenylazo)aniline. | ||||
| 1 | 10 | 720 ± 50 | 110 | 478; 33000 |
| 2 | 11 | 670 ± 50 | 80 | 490; 30000 |
| 3 | 12 | 810 ± 50 | 110 | 484; 31500 |
| 4 | DR1 a | 930 ± 50 | 90 | 488; 29500 |
Before measuring their EO properties, polymers were spin-coated into good quality films from a solution of 250 g L−1 in ortho-dichlorobenzene. Except polymer P4 which is poorly soluble, the other polymers P1–P3 display high solubility allowing to produce films with high optical quality (no light scattering) and with thicknesses up to 3.7 µm. These features represent practical advantages for the application. For each material, wire poling under high electric field (3.9 kV) was performed at 90 °C, closed to their Tg, for one hour. Then the films of polymers P2 and P3 bearing an alkyl spacer could be cross-linked at 120 °C, whereas polymers P1 and P4 containing rigid aryl linkers need to be cured at 150 °C. Finally, films were rapidly cooled to room temperature to freeze the orientation of the chromophores, while the electric field was still on. The conversion of the cross-linking reaction was assessed by the intensity change of the azide stretching band at 2100 cm−1 directly recorded on the film. It is worthwhile to note that the reaction of the azides with the alkynes is close to 100% in polymers P1–P3 after 100 min at Tc, while in polymer P4 the conversion reached only 50% after 300 minutes at 150 °C. In polymer P4, the reactive groups are connected to the polymer chain and to the chromophorevia rigid spacers, resulting in a lower flexibility, hence a lower mobility. As a consequence, the collision between an azide and an alkyne becomes less frequent explaining the lower cross-linking rate and the lower conversion even under prolonged heating. Upon cross-linking, the polymer films became harder and they lost the solubility in THF. It should be pointed out that the rate of cross-linking is faster in the presence of the electric field than by simple heating. This observation is consistent with the observation that dipolar Huisgen cyclo-addition reaction is generally accelerated in polar media.37 Indeed, the strong electric field applied to the polymer film during the poling certainly modifies the dielectric constant of the material or increases the polarization of the azide and/or of the alkyne groups. The d33 electro-optic coefficients were measured at 1064 nm by second harmonic generation (SHG) and the results are gathered in Table 2. First of all, the electro-optic coefficients of the polymers P1–P3 are relatively similar and in the same range as that measured in DR1-grafted methacrylate polymer with a similar chromophore loading.10,38 Interestingly, polymer P4, which contains two rigid spacers to connect the azido and the alkyne groups displays a much lower electro-optic coefficient. We interpret this result as a lower poling efficiency owing to a segregation of the chromophores along the polymer chain resulting from the formation of a block polymer with the chromophores positioned aside next to one another. This is consistent with the much lower solubility of P4 compared to that of P1–P3. Indeed, the reactivity of monomer 16 is certainly much higher than that of 10, because we observed that 16 spontaneously homopolymerizes in the NMR tube after only a few hours at room temperature; besides, a prolonged storage in the freezer (within one week) results also in homopolymerization of 16 in the flask. Conversely, monomer 10 is very stable in the same conditions. As already reported in the literature, the properties of a chromophore assembly strongly depend on the relative interchromophoric distance, explaining the decrease of the macroscopic NLO response as the chromophore concentration in the polymer reaches a certain value.2,39
Finally, the thermal stabilities of the bulk NLO activities of the polymers were investigated by depoling experiments, in which the real time decay of the SHG signal was monitored as a function of the temperature (Fig. 5).
 | ||
| Fig. 5 Thermal stability of the studied polymers upon heating an initially poled film at a rate of 2 °C min−1 in the air. | ||
Several interesting conclusions can be drawn from Fig. 5. First, the cross-linking reaction improves the stability of the EO activity of polymers. For example, when the polymer P1 is simply poled but not cross-linked, the chromophores start to relax at 80 °C, which is close to the Tg in agreement with what is generally published with other polymers. Second, the stability of the chromophore alignment correlates with the flexibility of the spacers connecting the cross-linking groups. For example, if we compare P1 and P3, which essentially differ by the flexibility of the spacer between the azide and the chromophore, the stability is significantly lower with the floppy spacer in P3, since the decrease of the NLO activity starts at 110 °C in P3, which is about 30 °C lower than in P1. Third, the position of the reactive groups could be inverted without appreciable change on the stability and the poling efficiency as evidenced by the properties of polymers P2 and P3. In these two latter polymers the azide and alkyne groups were switched from the monomer side chain to the chromophore substituent (Fig. 1). However, the value of the electro-optic coefficient and its stability are very similar (Table 2 and Fig. 5). Finally, although the properties of polymer P4 are poor, the study of this material teaches us one important point. The rigidity of spacers on both reactive groups imparts low mobility of the latter, hence a low conversion of the cross-linking reaction. As a result, the hardening of the polymer matrix is insufficient and the stabilization of the chromophores is poor leading to a decrease of the electro-optic activity at a lower temperature than in P1, which contains fully floppy spacers at both sides. However, the SHG signal in P4 decreases less steeply as compared to the other systems, underlining that when the chromophores are cross-linked with fully rigid spacers their mobility is more reduced than with flexible ones. In conclusion, although the stability is conditioned by the rigidity of the spacers, a certain degree of mobility must be preserved to ensure a high yield of the cross-linking process at a temperature close to the Tg.
Conclusions
In this work, three new polymers exhibiting structural variations were prepared and their NLO properties were fully characterized. Preliminary kinetic studies of the cross-linking reaction were undertaken in solution and demonstrated that it is possible to adjust the cross-linking temperature by changing the nature of the substituent on the alkyne group. Therefore, this new cross-linking reaction could certainly be advantageously extended to a number of polymers with different Tgs. The analysis of different substituents indicates that the trimethylsilyl group is certainly one of the most suitable groups to be used with methacrylate polymers because of its high synthetic accessibility, its ability to solubilize the polymer, and its reactivity with azide, which is relatively close to that measured with a terminal alkyne. The reactivity ratios of monomers 10 and 13 were also determined and contrary to our expectations, it was found that the monomer connected to the DR1 chromophore (10) is more reactive than trimethylsilyl-propargyl methacrylate 13. However, when the molar ratio of the chromophore is not too high, the chromophores are relatively diluted within the propargyl units. Another interesting finding of this study is that the stability and the poling efficiency of the NLO properties do not depend on the position of the reactive groups (azide or trimethylsilylacetylated groups) in the polymer, since they are interchangeable (on the chromophore or as polymer side chain). This feature can be useful, because it offers to the chemists the possibility to introduce, on the chromophore, either the azide or the alkyne group depending on their easiest synthetic accessibility. This structural versatility is all the more interesting to extend this cross-linking strategy to more synthetically demanding chromophores which exhibit high quadratic hyperpolarizability. Finally, this study highlights the importance of the rigidity of the spacers, connecting the reactive groups, on both the poling efficiency and on the stability of the NLO bulk property. Indeed, NLO activity of the materials bearing one rigid aryl linker is maintained up to 137 °C, while the other polymers with flexible alkyl linkers start to lose the macroscopic NLO activity at 110 °C. On the other hand, polymer P4, containing rigid spacers to link both reactive groups, exhibits a sluggish cross-linking reaction and therefore a low cross-linking conversion. Both of these aspects make it the less interesting material within this series. In conclusion, while the rigidity of the spacers is certainly critical to ensure a high stability of the orientation of the chromophores, one should introduce a certain degree of flexibility in the system to enable sufficient mobility of the cross-linking groups to ensure an efficient poling and a rapid cross-linking reaction.Acknowledgements
Agence Nationale de la Recherche (ANR-Télécom with project ModPol) and Région Pays de la Loire (MILES-MATTADOR program) are gratefully acknowledged for the financial support of these researches. V.R. thanks F. Adamietz for technical assistance and the Région Aquitaine for financial support in optical, laser, and computer equipment. We thank Dr Pierre-Antoine Bonnardel from PCAS company for large scale synthesis of the monomers 10 and 13.References
- J. Luo, X.-H. Zhou and A. K. Y. Jen, J. Mater. Chem., 2009, 19, 7410–7424 RSC.
- P. A. Sullivan and L. R. Dalton, Acc. Chem. Res., 2010, 43, 10–18 CrossRef CAS.
- H. Saadeh, D. Yu, L. M. Wang and L. P. Yu, J. Mater. Chem., 1999, 9, 1865–1873 RSC.
- O. Ostroverkhova and W. E. Moerner, Chem. Rev., 2004, 104, 3267–3314 CrossRef CAS; S. R. Marder, B. Kippelen, A. K. Y. Jen and N. Peyghambarian, Nature, 1997, 388, 845–851 CrossRef CAS.
- D. M. Burland, R. D. Miller and C. A. Walsh, Chem. Rev., 1994, 94, 31–75 CrossRef CAS.
- M. H. Davey, V. Y. Lee, L. M. Wu, C. R. Moylan, W. Volksen, A. Knoesen, R. D. Miller and T. J. Marks, Chem. Mater., 2000, 12, 1679–1693 CrossRef CAS.
- Y. Enami, C. T. Derose, D. Mathine, C. Loychik, C. Greenlee, R. A. Norwood, T. D. Kim, J. Luo, Y. Tian, A. K. Y. Jen and N. Peyghambarian, Nat. Photonics, 2007, 1, 180–185 Search PubMed; H. Ma, S. Liu, J. Luo, S. Suresh, L. Liu, S. H. Kang, M. Haller, T. Sassa, L. R. Dalton and A. K. Y. Jen, Adv. Funct. Mater., 2002, 12, 565–574 CrossRef CAS; M. Haller, J. Luo, H. Li, T.-D. Kim, Y. Liao, B. H. Robinson, L. R. Dalton and A. K. Y. Jen, Macromolecules, 2004, 37, 688–690 CrossRef CAS; J. Luo, M. Haller, H. Li, T.-D. Kim and A. K. Y. Jen, Adv. Mater., 2003, 15, 1635–1638 CrossRef CAS.
- H. Ma, B. Chen, T. Sassa, L. R. Dalton and A. K. Y. Jen, J. Am. Chem. Soc., 2001, 123, 986–987 CrossRef CAS; J. Luo, S. Liu, M. Haller, L. Liu, H. Ma and A. K. Y. Jen, Adv. Mater., 2002, 14, 1763–1768 CrossRef CAS; J. Luo, M. Haller, H. Ma, S. Liu, T.-D. Kim, Y. Tian, B. Chen, S.-H. Jang, L. R. Dalton and A. K. Y. Jen, J. Phys. Chem. B, 2004, 108, 8523–8530 CrossRef CAS.
- Y. Bai, N. Song, J. P. Gao, X. Sun, X. Wang, G. Yu and Z. Y. Wang, J. Am. Chem. Soc., 2005, 127, 2060–2061 CrossRef CAS.
- D. Bosc, F. Foll, B. Boutevin and A. Rousseau, J. Appl. Polym. Sci., 1999, 74, 974–982 CrossRef CAS.
- C. Monnereau, E. Blart, B. Illien, M. Paris and F. Odobel, J. Phys. Org. Chem., 2005, 18, 1050–1058 CrossRef CAS; A. Apostoluk, J. M. Nunzi, V. Boucher, A. Essahlaoui, R. Seveno, H. W. Gundel, C. Monnereau, E. Blart and F. Odobel, Opt. Commun., 2006, 260, 708–711 CrossRef CAS; C. Monnereau, E. Blart, V. Montembault, L. Fontaine and F. Odobel, Tetrahedron, 2005, 61, 10113 CrossRef CAS.
- A. Scarpaci, E. Blart, V. Montembault, L. Fontaine, V. Rodriguez and F. Odobel, Chem. Commun., 2009, 1825–1827 RSC.
- W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2007, 28, 15–54 CrossRef CAS; D. Fournier, R. Hoogenboom and U. S. Schubert, Chem. Soc. Rev., 2007, 36, 1369–1380 RSC.
- M. A. White, A. Maliakal, N. J. Turro and J. Koberstein, Macromol. Rapid Commun., 2008, 29, 1544–1548 CrossRef CAS.
- V. Bock, D. H. Hiemstra and J. H. van Maarseveen, Eur. J. Org. Chem., 2006, 51–68 CrossRef CAS.
- D. D. Diaz, S. Punna, P. Holzer, A. K. McPherson, K. B. Sharpless, V. V. Fokin and M. G. Finn, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 4392–4403 CrossRef CAS.
- Z. Li, Q. Zeng, G. Yu, Z. Li, C. Ye, Y. Liu and J. Qin, Macromol. Rapid Commun., 2008, 29, 136–141 CrossRef CAS.
- A. Scarpaci, E. Blart, V. Montembault, L. Fontaine, V. Rodriguez and F. Odobel, ACS Appl. Mater. Interfaces, 2009, 1, 1799–1806 Search PubMed; Z. Li, Q. Zeng, G. Yu, Z. Li, C. Ye, Y. Liu and J. Qin, Macromol. Rapid Commun., 2008, 29, 136–141 CrossRef CAS; L. Nurmi, J. Lindqvist, R. Randev, J. Syrett and D. M. Haddleton, Chem. Commun., 2009, 2727–2729 RSC.
- A. Scarpaci, C. Cabanétos, E. Blart, V. Montembault, L. Fontaine, V. Rodriguez and F. Odobel, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5652–5660 CrossRef CAS.
- H. L. Cohen, J. Polym. Sci., Polym. Chem. Ed., 1981, 19, 3269–3284 CrossRef CAS; H. L. Cohen, J. Polym. Sci., Polym. Chem. Ed., 1981, 19, 1337–1347 CrossRef CAS.
- J. A. Codelli, J. M. Baskin, N. J. Agard and C. R. Bertozzi, J. Am. Chem. Soc., 2008, 130, 11486–11493 CrossRef CAS; J. A. Johnson, J. M. Baskin, C. R. Bertozzi, J. T. Koberstein and N. J. Turro, Chem. Commun., 2008, 3064–3066 RSC; J. M. Baskin, J. A. Prescher, S. T. Laughlin, N. J. Agard, P. V. Chang, I. A. Miller, A. Lo, J. A. Codelli and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 16793–16797 CrossRef CAS.
- M. Kunishima, C. Kawachi, J. Monta, K. Terao, F. Iwasaki and S. Tani, Tetrahedron, 1999, 55, 13159 CrossRef CAS.
- B. S. Sumerlin, N. V. Tsarevsky, G. Louche, R. Y. Lee and K. Matyjaszewski, Macromolecules, 2005, 38, 7540–7545 CrossRef CAS.
- Y. Fan, Y.-M. Zhu, F.-R. Dai, L.-Y. Zhang and Z.-N. Chen, Dalton Trans., 2007, 3885–3892 RSC.
- L. Sanguinet, J.-L. Pozzo, V. Rodriguez, F. Adamietz, F. Castet, L. Ducasse and B. Champagne, J. Phys. Chem. B, 2005, 109, 11139–11150 CrossRef CAS.
- T. Verbiest, K. Clays and V. Rodriguez, Second-Order Nonlinear Optical Characterization Techniques: an Introduction, CRC Press, New York, 2009 Search PubMed.
- V. Rodriguez, F. Adamietz, L. Sanguinet, T. Buffeteau and C. Sourisseau, J. Phys. Chem. B, 2003, 107, 9736–9743 CrossRef CAS.
- V. Rodriguez, J. Chem. Phys., 2008, 128, 064707/064701–064707/064710; V. Rodriguez and C. Sourisseau, J. Opt. Soc. Am. B, 2002, B19, 2650–2664 Search PubMed.
- W. Lwowski, 1,3-Dipolar Cycloaddition Chemistry, Wiley-Interscience, New York, 1984 Search PubMed.
- V. Ladmiral, G. Mantovani, G. J. Clarkson, S. Cauet, J. L. Irwin and D. M. Haddleton, J. Am. Chem. Soc., 2006, 128, 4823–4830 CrossRef CAS; B. Helms, J. L. Mynar, C. J. Hawker and J. M. J. Frechet, J. Am. Chem. Soc., 2004, 126, 15020–15021 CrossRef CAS.
- M. Fineman and S. D. Ross, J. Polym. Sci., 1950, 5, 259–262 CrossRef CAS.
- J. P. Kennedy, T. Kelen and F. Tüdös, J. Polym. Sci., Polym. Chem. Ed., 1975, 13, 2277–2289 CrossRef CAS.
- J. C. Brosse, J. M. Gauthier and J. C. Lenain, Makromol. Chem., 1983, 184, 505–517 CrossRef CAS.
- M. Ergin, B. Kiskan, B. Gacal and Y. Yagci, Macromolecules, 2007, 40, 4724–4727 CrossRef CAS.
- G. L'Abbe, Chem. Rev., 1969, 69, 345–363 CrossRef CAS.
- J. L. Oudar and D. S. Chemla, J. Chem. Phys., 1977, 66, 2664–2668 CrossRef CAS.
- Z.-X. Wang and H.-L. Qin, Chem. Commun., 2003, 2450–2451 RSC; N. Butler Richard, J. Cunningham William, G. Coyne Anthony and A. Burke Luke, J. Am. Chem. Soc., 2004, 126, 11923–11929 CrossRef CAS.
- S. K. Yesodha, C. K. Sadashiva Pillai and N. Tsutsumi, Prog. Polym. Sci., 2004, 29, 45–74 CrossRef CAS.
- V. P. Yuriy, V. P. Oleg and R. D. Larry, ChemPhysChem, 2004, 5, 1821–1830 CrossRef CAS; F. Terenziani, S. Ghosh, A.-C. Robin, P. K. Das and M. Blanchard-Desce, J. Phys. Chem. B, 2008, 112, 11498–11505 CrossRef CAS.
Footnote |
| † Both authors equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2011 |