Preparation of hydrophobic helical poly(N-propargylamide)s in aqueous medium via a monomer/cyclodextrin inclusion complex
Lei
Ding
ab,
Yi
Li
ab,
Jianping
Deng
*ab and
Wantai
Yang
*ab
aState Key Laboratory of Chemical Resource Engineering, Beijing University of Chemical Technology, Beijing, 100029, China. E-mail: dengjp@mail.buct.edu.cn; Fax: +86-10-6443-5128; Tel: +86-10-6443-5128yangwt@mail.buct.edu.cn
bCollege of Materials Science and Engineering, Beijing University of Chemical Technology, Beijing, 100029, China
First published on 11th November 2010
Abstract
We present a facile approach for the preparation of hydrophobic helical poly(N-propargylamide)s in aqueous medium instead of toxic and volatile organic solvent by using a monomer/cyclodextrin inclusion complex. Four hydrophobic substituted acetylene monomers were investigated in this study. Their inclusion complexes with hydroxypropyl-β-cyclodextrin (HP-β-CD) and hydroxypropyl-γ-cyclodextrin (HP-γ-CD) were prepared in water and identified with FT-IR and NMR spectroscopy. Polymerizations of the complexes were successfully carried out in aqueous solution in the presence of a water-soluble Rh-based catalyst [Rh(cod)2BF4; cod = 1,5-cyclooctadiene]. The as-prepared poly(N-propargylamide)s exhibited little difference in composition from the counterparts obtained in organic solvent according to FT-IR analysis. Circular dichroism and UV-Vis absorption spectra demonstrated that the as-prepared poly(N-propargylamide)s could take ordered helical conformations. The reusability of cyclodextrins in the polymerization was investigated quantitatively, which should be one of cyclodextrins' advantages but has never been highlighted before in literature. The versatile method for preparing hydrophobic helical poly(N-propargylamide)s can efficiently reduce the use of noxious organic solvents and can be applicable to the preparation of other helical polymers.
Introduction
The helix is one of the most ubiquitous structures in biomacromolecules, e.g., proteins and DNA.1,2 Synthetic polymers able to form helixes also attract particular interest from polymer chemists owing to their unique structures and potential applications. Particularly in the past two decades, the progress in polymer synthesis has enabled the design and preparation of novel polymers with well-ordered helical structures, e.g., polymethacrylates,3 polyisocyanides,4 polyisocyanates,5 polysilanes,6 and polyacetylenes.7–9 Among these synthetic helical polymers, polyacetylenes have played an increasingly important role as typical conjugated polymers. Many polymer chemists have made significant contributions towards the progress in helical polyacetylenes. For example, Yashima et al.,7 Tang and co-workers,8 Nakano and Okamoto,1 and Masuda and co-workers9 found that polyacetylenes with appropriate substituents can adopt helical conformations under suited conditions. We have also prepared three categories of helical polyacetylenes, including poly(N-propargylamide)s,10–12 poly(N-propargylsulfamide)s,13 and poly(N-propargylurea)s.14 We further prepared stable emulsions consisting of helical polymers from substituted acetylene monomers,15,16 novel core (helical polyacetylenes)/shell (vinyl polymers) strucutred nanoparticles,17vinyl polymer hollow particles grafted with helical polymer chains,18 and optically active emulsions from achiral substituted acetylene monomers in chiral micelles by asymmetric polymerization.19 Although Tang et al.20 and Yashima and co-workers21 have reported the successful synthesis of helical polyacetylenes in water, most of the aforementioned helical polymers were prepared in toxic and volatile organic solvents rather than harmless aqueous medium.The importance of using water as medium for chemical reactions is ever-increasing as a way to environmentally benign events.22,23Water is thus considered as the favored solvent in chemistry and also in polymer research field.24–27Polymerizations in aqueous medium are recently drawing growing attention owing to the advantages of using water as solvent. Many reports have been found to deal with the aqueous radical polymerization of vinyl monomers.26–29 In particular, aqueous catalytic polymerizations, which combine the advantages of relatively easy control of the microstructure of the polymersviacatalytic polymerization and the benefits of using water as polymerization solvent, are gathering continuously increasing interest.25
In order to prepare hydrophobic helical poly(N-propargylamide)s in aqueous medium, we created a facile approach via polymerizing the monomer/cyclodextrin inclusion complex in water.30Cyclodextrins (CDs), as cyclic oligosaccharides consisting of 6 (α), 7 (β), or 8 (γ) glucopyranose units linked by 1,4-α-glucosidic bonds,31,32 have been widely applied in polymer chemistry such as for the preparation of supramolecular polyrotaxanes,33 chemical sensors,34 supramolecular self-assemblies,35 hydrogels,36 and nanogels.37 CDs have also been extensively used in click chemistry and the related reactions,38,39 and attracted much attention as aqueous-based hosts for chiral recognition40 and chiral separations.41 CDs' unique structures of a polar hydrophilic outer shell and relatively hydrophobic cavity are in particular interesting to polymer chemists because CDs can include a suitable guest to form a host/guest inclusion complex, by which hydrophobic monomers can undergo polymerizations in aqueous medium instead of noxious organic solvents. The group of Ritter has reported the first study using CD during the formation of different polymers in aqueous solution and made significant contributions in this interesting research field.42–46
In our previous research, CDs were used to prepare CD/GMA47 and CD/MMA inclusion complexes,48 with which novel hydrogels were successfully obtained and UV-induced polymerizations were smoothly carried out, respectively. We also reported the first catalytic polymerizations in aqueous medium by using monomer/cyclodextrin inclusion complex.30 However, some monomers with large phenyl pendent groups cannot be prepared viaHP-β-CD/monomer inclusion complex. In the present article, we successfully circumvent this problem by employing HP-γ-CD. It should be stressed that this is the first attempt in the literature to use γ-CD/monomer inclusion complex in catalytic polymerizations, although Uyar et al. reported the radical polymerization of styrene in γ-CD channels.49 In addition, HP-β-CD and HP-γ-CD were used in this work because their solubility in water was much better than that of β-CD and γ-CD. We further found that the poly(N-propargylamide)s separately obtained via two different routes (polymerization in aqueous media by using monomer inclusion complex vs.polymerization in organic solvent by directly using monomer) exhibited no pronounced difference in terms of polymers' yield, composition, and the ability to form helical conformations. Also of particular importance is that cyclodextrin used in the polymerization system can be reused several times, further demonstrating that cyclodextrins can be used as an environment-friendly material.
Experimental section
Materials
Hydroxypropyl-β-cyclodextrin (HP-β-CD) was purchased from Acros, with an average degree of substitution about 1.0 per glucose unit mainly in 2-position. Hydroxypropyl-γ-cyclodextrin (HP-γ-CD) was purchased from Aldrich, with an average degree of substitution about 0.6 per glucose unit mainly in 2-position. Monomers were synthesized according to earlier reports (monomer 1, ref. 10; monomer 2, ref. 11; monomer 3, ref. 9; and monomer 4, ref. 12). [Rh(cod)2BF4; cod = 1,5-cyclooctadiene] was obtained from Aldrich; [Rh(nbd)B(C6H5)4; nbd = norbornadiene] was synthesized according to ref. 50Water was freshly deionized before use. THF (tetrahydrofuran) and CHCl3 were purified by distilling under reduced pressure. All other reagents were used as received.Measurements
1H and 13C NMR spectra were obtained on a Bruker AV600 spectrometer at room temperature. For CDs and monomer/CD complexes, the NMR spectra were recorded in D2O; for monomers, CDCl3 was used as solvent. FTIR spectra were recorded with a Nicolet NEXUS 670 spectrophotometer. Circular dichroism and UV-Vis absorption spectra were recorded on a JASCO J - 810 spectrophotometer. Number average molecular weights (Mn) and molecular weight distributions (Mw/Mn) of polymers were determined by GPC (Waters 515–2410 system) calibrated by using polystyrenes as standards and THF as eluent.Preparation of monomer/CD inclusion complex
Taking monomer 4/HP-γ-CD system (Table 1) as an example, the typical procedure for preparing monomer/CD inclusion complex is as follows. HP-γ-CD (0.316 g, 0.2 mmol) was dissolved in 5 ml deionized water. To this solution dropwise added monomer 4 (0.02 g, 0.1 mmol)/THF (0.1 ml) solution under constant stirring. The mixture was stirred for 2 h at room temperature, giving rise to clear solution without any precipitate. Water was then removed using a rotaevaporator and the acquired inclusion complex in the form of a white solid was dried for 24 h at 50 °C under vacuum.| Monomer | HP-β-CD | HP-γ-CD | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry |
|
Appearance of the complex in watera | Inclusion complex or not | Appearance of the complex in watera | Inclusion complex or not | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a “Clear” means that no free monomer was left and “turbid” means that only part of the monomer was included by CD under the investigation conditions. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 |
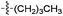
|
Clear | Yes | Clear | Yes | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 |

|
Clear | Yes | Clear | Yes | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 |

|
Clear | Yes | Clear | Yes | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 |

|
Turbid | No | Clear | Yes | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||

Polymerization of monomer/CD inclusion complex in water
A typical polymerization procedure is as follows, taking monomer 4/HP-γ-CD inclusion complex as a representative example. Polymerization was carried out in water under nitrogen atmosphere at a predetermined temperature, with monomer 4/HP-γ-CD inclusion complex of 1.68 g (containing monomer 4, 0.1 g, 0.5 mmol) and catalyst (e.g., [Rh(cod)2BF4], 0.002 g, 0.005 mmol). After polymerization for a predetermined period of time, the resulting solution containing precipitate was poured into a large amount of water. The solid polymer was collected by filtration and washed with water and ethyl acetate three times respectively to exclude the residual HP-γ-CD and monomer. The product was then dried under reduced pressure.Polymerization of the pure monomer in chloroform
Taking monomer 4 as an example, the major procedure is stated below. Polymerization of the pure monomer 4 was carried out with [Rh(nbd)B(C6H5)4] as a catalyst in chloroform at 30 °C for 3 h under the following conditions: [monomer] = 0.1 mol L−1, [catalyst] = 10 mmol L−1. After polymerization, the resultant solution was poured into a large amount of n-hexane to precipitate the formed polymer. The precipitate was filtered and then dried under reduced pressure.Investigation of the reusability of CD in the polymerization system
Herein we take monomer 2/HP-β-CD system as a representative for the investigation of CD's reusability in polymerization systems. The polymerization procedure is the same as stated above. After polymerization, the polymer was collected by filtration and washed with deionized water five times. The filtrates of each step were collected and then evaporated using a rotaevaporator under reduced pressure. The obtained solid was dried in vacuum until the mass reached a constant value. Afterwards, the solid was weighed and recorded as mcd (the mass of the HP-β-CD remaining in the solution after polymerization).We define three parameters as the following to investigate the reusability of HP-β-CD quantitatively. The total conversion (X) of monomer 2 is defined as
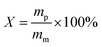 | (1) |
 | (2) |
 | (3) |
Results and discussion
Characterization of the monomer/CD inclusion complex
It is well known that the formation of inclusion complex between monomer and cyclodextrin depends on the size of the monomer, especially the size of the substituents (R groups in Table 1) on the monomer. In our previous report,30 a series of substituted acetylene monomers with different substituents were examined with regard to their abilities to form inclusion complex with HP-β-CD. Interestingly, only the monomers with a small substituent can form inclusion complex with HP-β-CD; and those bearing large phenyl groups cannot form inclusion complex. In this paper, four monomers with varied substituents were investigated. The chemical structures of the four investigated monomers are listed in Table 1. All the monomers with the exception of monomer 4 can form inclusion complex with both HP-β-CD and HP-γ-CD; for monomer 4, it only formed inclusion complex with HP-γ-CD. It seems that monomer 4 and the cavity of HP-γ-CD rather than HP-β-CD are in good size match.The formation of an inclusion complex was preliminarily proved by observing the change in the appearance of the involved aqueous solution. When the solution of monomer 4/THF was added into the HP-γ-CD aqueous solution, the monomer immediately precipitated and dispersed in the system. Nevertheless after stirring at room temperature for about 30 min, the monomer precipitate disappeared and a clear, homogenous solution was formed. For a further confirmation of the occurrence of inclusion, NMR and FTIR spectroscopy techniques were employed, both of which have been proved effective for the confirmation of inclusion complex.47,48,51,52 The relevant results are presented in Fig. 1 (NMR) and Fig. 2 (FTIR), and will be discussed in detail later.
 | ||
| Fig. 1 (A) 1H NMR spectra of a: HP-γ-CD; b: monomer 4/HP-γ-CD inclusion complex; and c: monomer 4. (B) 13C NMR spectra of d: HP-γ-CD; e: monomer 4/HP-γ-CD inclusion complex; and f: monomer 4. The spectra of HP-γ-CD and monomer 4/HP-γ-CD were recorded in D2O while the spectrum of monomer 4 was acquired in CHCl3. | ||
 | ||
| Fig. 2 FTIR spectra of a: HP-γ-CD; b: monomer 4/HP-γ-CD inclusion complex; and c: monomer 4 (KBr tablet). | ||
A comparison of the 1H and 13C NMR spectra of the monomer 4/HP-γ-CD inclusion complex and those of pure monomer 4 and pure HP-γ-CD is presented in Fig. 1. Table 2 and Table 3 illustrate the corresponding chemical shifts of monomer 4 observed in Fig. 1. The data in Table 2 show that all the corresponding signals of the protons in monomer 4 shifted downfield after the inclusion with the exception of the proton in the –NH– group. Furthermore, no new peaks were observed after the inclusion and only chemical shifts occurred to the protons, indicating that the complexation was a physical process rather than a chemical reaction. In order to investigate the inclusion mode of monomer 4/HP-γ-CD inclusion complex, we further measured 13C NMR spectra of the pure HP-γ-CD, the pure monomer 4, and their inclusion complex. The results are presented in Fig. 1B and the detailed data are listed in Table 3. For the guest monomer 4, C (1–3) and C (6–11) showed downfield shifts, while only C–5 (carbon in the carbonyl group) shifted in the opposite direction. This is in good agreement with the observations in the 1H NMR spectra above, namely, all the protons shifted downfield except the proton in the amide group. A combination of the above results clearly demonstrates that in monomer 4, atoms C–1 to C–11, except C–5, are in the same environment after the formation of inclusion complex. Therefore, the inclusion complex was formed in a mode as schematically presented in Scheme 1. This inclusion complex mode is similar to that already found in the glycidyl methacrylate (GMA/HP-β-CD) system.47
| Proton | δ free | δ complex | Δδ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a The data were based on Fig. 1A in the text. The spectrum of monomer 4/HP-γ-CD was recorded in D2O while the spectrum of monomer 4 was acquired in CHCl3. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| H-1 | 2.134 | 1.960 | −0.174 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| H-3 | 3.944 | 3.818 | −0.126 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| H-4 | 5.347 | 5.549 | +0.202 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| H-7 | 1.567 | 1.427 | −0.140 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| H-9 | 7.285 | 7.257 | −0.028 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| H-10 | 7.369 | 7.330 | −0.039 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| H-11 | 7.255 | 7.167 | −0.088 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Carbon | δ free | δ complex | Δδ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a The data were based on Fig. 1B in the text. The spectrum of monomer 4/HP-γ-CD was recorded in D2O while the spectrum of monomer 4 was acquired in CHCl3. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| C-1 | 71.30 | 69.03 | −2.27 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| C-2 | 79.55 | 78.03 | −1.52 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| C-3 | 29.44 | 29.19 | −0.25 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| C-5 | 177.03 | 179.21 | +2.18 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| C-6 | 46.90 | 46.64 | −0.26 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| C-7 | 26.96 | 26.72 | −0.24 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| C-8 | 144.88 | 144.67 | −0.21 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| C-9 | 128.75 | 128.70 | −0.05 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| C-10 | 127.11 | 127.10 | −0.01 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| C-11 | 126.38 | 125.72 | −0.66 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 | ||
| Scheme 1 A schematic illustration of the monomer 4/HP-γ-CD inclusion complex. | ||
A further comparison between the 1H NMR spectrum of the monomer 4/HP-γ-CD inclusion complex (spectrum b, Fig. 1) and that of pure HP-γ-CD (spectrum a) demonstrates that all the corresponding signals of HP-γ-CD slightly shifted downfield in the presence of monomer 4. In theory, the signals at δ = 5.27, 5.07 and 3.97 ppm, which correspond to protons outside the cavity, should shift upfield. The detailed reason for this phenomenon is not yet clear at present. However, in the 13C NMR spectra, the carbons inside the cavity of HP-γ-CD shifted downfield while the carbons outside the cavity (δ = 101– 98, and 73–71 ppm) shifted upfield after inclusion occurrence, which is well in agreement with the result of monomer 4.
Besides NMR spectroscopy, FTIR spectroscopy measurements also provided powerful evidence for the formation of inclusion complex, as illustrated in Fig. 2. When compared to the spectra of the pure HP-γ-CD (spectrum a in Fig. 2) and monomer 4 (spectrum c), the characteristic bands of monomer 4 at 3321, 3286, 1649, and 1524 cm−1 all appeared in the spectrum of the monomer 4/HP-γ-CD inclusion complex (spectrum b) and the characteristic peaks of the cyclodextrin at 3395 and 1148–1027 cm−1 were also observed there. These observations indicated the formation of the expected inclusion complex. It should be pointed out here that before the measurement of FTIR spectroscopy, the free monomer in the inclusion complex was entirely excluded.
Polymerizations
All the four monomers underwent polymerizationsvia the two diverse routes depicted in Scheme 2 to provide deep insights into the polymerization of hydrophobic monomers in aqueous media. Taking monomer 4 as a typical example, polymerization of monomer 4/HP-γ-CD inclusion complex was carried out in water in the presence of hydrophilic Rh-based catalyst, [Rh(cod)2BF4], since hydrophobic Rh catalyst was found to be not effective.30 In contrast, polymerization of pure monomer 4 was conducted in chloroform in the presence of a hydrophobic Rh-based catalyst, [Rh(nbd)B(C6H5)4]. The detailed results and discussion will be presented below. | ||
| Scheme 2 Preparation and polymerization of monomer 4/HP-γ-CD inclusion complex in water in comparison to polymerization of uncomplexed monomer 4 in chloroform. | ||
Some earlier investigations demonstrated that,42–46 once the monomer in a monomer/cyclodextrin inclusion complex forms a polymer through radical polymerization, the resulting polymer frequently breaks away from the cyclodextrin and precipitates out from the solution. Hence a pure polymer can be obtained just by a simple filtration. This is also true in our catalytic polymerizations of hydrophobic substituted acetylene monomers. The resultant polymers were isolated by a facile filtration, and washed with water and then ethyl acetate to remove the free cyclodextrins and the residual monomer, respectively. After re-precipitation from THF into n-hexane, the as-prepared polymer was subjected to GPC and the results are listed in Table 4. From Table 4, although the number average molecular weights (Mns) of the polymers prepared in aqueous medium were a bit lower than those of their counterparts obtained in organic solvent, no pronounced difference can be observed in the yields of the polymers. Just as aforementioned, in the present aqueous polymerization systems, the monomer/cyclodextrin inclusion complexes were water-soluble but the as-prepared polymers were water- insoluble. Therefore, in principle the polymers should break away from the cavity of cyclodextrins and then precipitated, resulting in the termination of the polymer chains propagation. As a consequence, the molecular weights of the polymers are relatively low.
| Entry | Monomer | Cyclodextrin | Solvent | Yield (%) | M n b | M w/Mnb | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a The concentration of monomer was 0.1 mol L−1; the concentration of catalyst ([Rh(cod)2BF4]) was 1.0 mmol L−1; the polymerization temperature was 30 °C; and the polymerization time was 3 h. b Determined by GPC, using polystyrene as the standard and THF as eluent. c Cyclodextrin was not used, and the catalyst used was [Rh(nbd)B(C6H5)4]. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | 1 | HP-β-CD | H2O | 97 | 4600 | 1.22 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | 1 | /c | CHCl3 | 98 | 9000 | 1.69 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | 2 | HP-β-CD | H2O | 97 | 6200 | 1.93 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | 2 | /c | CHCl3 | 95 | 9000 | 2.35 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | 3 | HP-β-CD | H2O | 94 | 4500 | 1.18 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | 3 | /c | CHCl3 | 96 | 6500 | 2.41 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | 4 | HP-γ-CD | H2O | 96 | 4000 | 1.13 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 | 4 | /c | CHCl3 | 97 | 4200 | 3.71 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
To further clarify the assumption that the polymers prepared via aqueous polymerization fell off the cyclodextrins during polymerization, the polymers prepared by the two different approaches (cf.Scheme 2) were subjected to FTIR measurement. The relevant results were presented in Fig. 3, where two spectra (spectra a and b: for polymer 4 prepared in aqueous medium and in organic solvent, respectively) are shown simultaneously for a visually clear comparison. It can be seen that the two spectra were almost the same in the characteristic bands of the two polymers. The peaks appeared at 3356, 1647, and 1515 cm−1 were assigned to the amine groups and amide groups in the side chains of the polymers. More importantly, hydroxyl group (–OH) was hardly observed in the FTIR spectrum of polymer 4 obtained in aqueous solution, indicating that there was no cyclodextrin molecules threaded on the polymer chains. It is worthwhile to point out that polymer 4 prepared via aqueous polymerization showed little difference in solubility in organic solvent from the polymer 4 prepared in organic solvent. This is also in well accordance with the fact that HP-γ-CD broke away from the polymer chains during polymerization; otherwise the residual HP-γ-CD would adversely affect the solubility of the polymer.
 | ||
| Fig. 3 FTIR spectra of a: polymer 4 prepared by using monomer 4/HP-γ-CD inclusion complex in water; and b: polymer 4 prepared in chloroform (KBr tablet). | ||
Helical structure of the polymers
The previous studies10–19 showed that polymerizations in the presence of Rh-based catalyst provided high-yield polymers with cis contents of approximately 100%. It also has been known that all the four polymers except polymer 1 (Table 1) when prepared in organic solvents, could adopt a helical structure under suited conditions.9–12 Moreover, monomer 3 contains a chiral center and shows intense circular dichroism effects. But there is no circular dichroism signal observed in polymer 2 and polymer 4 because they have no chiral center. The secondary structures of the resulting polymers in this study were investigated by UV-Vis and circular dichroism spectroscopy analysis, which have been widely used for exploring the secondary structures of polymers.10–19 The relevant results are displayed in Fig. 4 and Fig. 5. | ||
| Fig. 4 A comparison of the UV-vis spectra of polymer 2 and polymer 4 prepared in water and in chloroform. For the polymerization conditions, cf.Table 4, entry 3 and 4 (polymer 2); entry 7 and 8 (polymer 4). | ||
 | ||
| Fig. 5 A comparison of A: UV-vis absorption spectra and B: circular dichroism spectra of polymer 3 prepared in water and polymer 3 prepared in chloroform. For the polymerization conditions, cf.Table 4, entry 5 and 6. | ||
Fig. 4 shows the UV-Vis spectra of the two polymers from monomer 2 and monomer 4 (for polymer 2, entry 3, in water; entry 4, in chloroform in Table 4. For polymer 4, entry 7, in water; and entry 8, in chloroform in Table 4). All the spectra showed a strong UV-vis absorption at about 390 nm. Although the absorption strength of the polymers prepared in water was not the same as that of the counterparts obtained in chloroform, little difference was observed in the spectra in terms of profile and the maximum wavelength. This indicates that polymerization in aqueous medium can also allow the polymers to form helical structures. The UV-Vis absorption spectra and circular dichroism effects of the two polymers derived from monomer 3 (entry 5, in water; and entry 6, in chloroform in Table 4) were displayed in Fig. 5. Similar to the observations in polymer 2 and 4, the two spectra in Fig. 5 were almost the same in their profiles either in their UV-Vis spectra or circular dichroism spectra. Monomer 3 has a chiral center and thus a circular dichroism signal can be seen in the circular dichroism spectra of polymer 3. Polymer 1 prepared by using a monomer 1/HP-β-CD inclusion complex did not show any absorption peaks in a wavelength range from 300 to 400 nm in both UV-Vis and circular dichroism spectra because polymer 1 cannot form a helical conformation under the examined conditions, as reported elsewhere.10 According to the above discussions, polymerizations by using monomer/cyclodextrin inclusion complexes can provide the expected polymers and did not affect the secondary structures of the thus-prepared polymers, irrespective of their ability or inability to adopt helical structure.
Investigation of the reusability of HP-β-CD
In this study, the reusability of the unthreaded CD in the polymerization system was particularly investigated. Since the polymerization system of monomer 2/HP-β-CD inclusion complex is much simpler than the other monomer systems, herein we take monomer 2/HP-β-CD as an example for the sake of clarity, and the experimental data are presented in Table 5. Interestingly, we found that in Table 5 when the concentration of HP-β-CD varied, there was no considerable difference in the yields of the polymers. All polymerizations provided high-yield (over 95%) polymers irrespective of the monomer 2/HP-β-CD molar ratios. In the first two entries in Table 5 where the amount of HP-β-CD was lower than that of monomer 2, the polymerizations still took place smoothly with little difference than the other entries. That is to say, HP-β-CD was initially not enough to form inclusion complexes with all the monomer molecules present in the system, but the final polymer yield was not reduced. This indicates that during polymerization, the HP-β-CD unthreaded from the polymer chains can form inclusion complex again with free monomer molecules. Namely, HP-β-CD can be used repeatedly for the formation of inclusion complex with monomer and thus the polymerization proceeded continuously until all the monomer was consumed.| HP-β-CD mmol | M2 mmol | Complex b mmol | X c % | X e c % | R CD c % | M n d | M w/Mnd | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a The concentration of the monomer was 0.1 mol L−1; the concentration of the catalyst ([Rh(cod)2BF4]) was 1.0 mmol L−1; the polymerization temperature was 30 °C; and the polymerization time was 3 h. b Theoretical quantity. c The total conversion, extra conversion, and HPCD surplus ratio were defined in the experimental part. d Determined by GPC, using polystyrene as the standard and THF as eluent. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 0.4 | 0.8 | 0.2 | 95.3 | 281.2 | 96.4 | 4700 | 1.18 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 0.8 | 0.8 | 0.4 | 97.7 | 95.4 | 96.0 | 3700 | 1.57 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 0.8 | 0.4 | 0.4 | 96.7 | 0 | 94.5 | 4200 | 1.28 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
This phenomenon can be easily explained by the extra conversion Xe. For example, when 0.4 mmol HP-β-CD and 0.8 mmol monomer 2 were used, 0.7624 mmol polymer 2 (based on the molecular weight of monomer 2) was obtained. Based on the mode of the monomer/HP-β-CD inclusion complex discussed above, only 0.2 mmol polymer 2 (based on the molecular weight of monomer 2; theoretical quantity) should be obtained. However, the extra conversion was 281.2% according to eqn (2). It thus can be further inferred that HP-β-CD was used repeatedly in the system based on the fact that the extra conversion was larger than 0; and in particular, the extra conversion was larger than 100% also further clearly demonstrates that HP-β-CD was used repeatedly more than once.
The concentration of the residual HP-β-CD after one batch of polymerization in the solution was also investigated. We found that in all the entries in Table 5, the HPCD surplus ratios were 90% and above, demonstrating that a majority of HP-β-CD was unthreaded from the polymer chain and remained in solution. This was also consistent with the results of the FTIR measurements in Fig. 2. Therefore, we conclude that most of the cyclodextrin used in the system can be reused in polymerization, and we believe that the present technique will find some practical applications in the synthesis of hydrophobic helical polymers.
Conclusions
Four substituted acetylene monomer/cyclodextrin inclusion complexes were prepared and identified by NMR and FTIR spectroscopy. One monomer molecule was found to be included into two cyclodextrin molecules. Polymerizations were readily conducted in aqueous medium by employing the monomer/cyclodextrin inclusion complexes. The as-prepared polymers exhibited little difference in yield and in composition from their counterparts obtained in organic solvents, although the number average molecular weights were a little lower for the former. The polymers prepared via aqueous polymerization can adopt helical conformations under the investigated conditions, similar to their counterparts obtained in organic solvents. Furthermore, the cyclodextrins can be reused in the polymerization. We believe that this aqueous catalytic polymerization technique based on a monomer/cyclodextrin inclusion complex, which is superior in terms of environmental protection to the traditional polymerizations in organic solvents, is promising for certain practical applications.Acknowledgements
This work was supported by the “Program for New Century Excellent Talents in University” (NCET-06-0096), “the National Science Foundation of China” (20974007), and the “Program for Changjiang Scholars and Innovative Research Team in University” (PCSIRT, IRT0706).Notes and references
- T. Nakano and Y. Okamoto, Chem. Rev., 2001, 101, 4013 CrossRef CAS.
- E. Yashima, K. Maeda, H. Iida, Y. Furusho and K. Nagai, Chem. Rev., 2009, 109, 6102 CrossRef CAS.
- T. Kawauchi, A. Kitaura, J. Kumaki, H. Kusanagi and E. Yashima, J. Am. Chem. Soc., 2008, 130, 11889 CrossRef CAS.
- J. G. Rudick and V. Percec, Acc. Chem. Res., 2008, 41, 1641 CrossRef CAS.
- S. K. Jha, K. S. Cheon, M. M. Green and J. V. Selinger, J. Am. Chem. Soc., 1999, 121, 1665 CrossRef CAS.
- M. Fujiki, Top. Curr. Chem., 2008, 284, 119 CAS.
- E. Yashima, K. Maeda and Y. Furusho, Acc. Chem. Res., 2008, 41, 1166 CrossRef CAS.
- J. E. Y. Lam and B. Z. Tang, Acc. Chem. Res., 2005, 38, 745 CrossRef CAS.
- J. Tabei, R. Nomura and T. Masuda, Macromolecules, 2002, 35, 5405 CrossRef CAS.
- J. P. Deng, J. Tabei, M. Shiotsuki, F. Sanda and T. Masuda, Macromolecules, 2004, 37, 1891 CrossRef CAS.
- J. P. Deng, J. Tabei, M. Shiotsuki, F. Sanda and T. Masuda, Macromolecules, 2004, 37, 5149 CrossRef CAS.
- J. P. Deng, J. Tabei, M. Shiotsuki, F. Sanda and T. Masuda, Macromolecules, 2004, 37, 7156 CrossRef CAS.
- Z. G. Zhang, J. P. Deng, W. G. Zhao, J. M. Wang and W. T. Yang, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 500 CrossRef CAS.
- J. P. Deng, X. F. Luo, W. G. Zhao and W. T. Yang, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 4112 CrossRef CAS.
- J. P. Deng, B. Chen, X. F. Luo and W. T. Yang, Macromolecules, 2009, 42, 933 CrossRef CAS.
- X. F. Luo, N. W. Kang, L. Li, J. P. Deng and W. T. Yang, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1661 CrossRef CAS.
- B. Chen, J. P. Deng, X. Q. Liu and W. T. Yang, Macromolecules, 2010, 43, 3177 CrossRef CAS.
- K. Zhou, L. Y. Tong, J. P. Deng and W. T. Yang, J. Mater. Chem., 2010, 20, 781 RSC.
- X. F. Luo, L. Li, J. P. Deng, T. T. Guo and W. T. Yang, Chem. Commun., 2010, 46, 2745 RSC.
- B. Z. Tang, W. H. Poon, S. M. Leung, W. H. Leung and H. Peng, Macromolecules, 1997, 30, 2209 CrossRef CAS.
- K. Maeda, H. Goto and E. Yashima, Macromolecules, 2001, 34, 1160 CrossRef CAS.
- U. M. Lindström, Chem. Rev., 2002, 102, 2751 CrossRef.
- R. Breslow, Acc. Chem. Res., 1991, 24, 159 CrossRef CAS.
- H. Ritter and M. Tabatabai, Prog. Polym. Sci., 2002, 27, 1713 CrossRef CAS.
- J. P. Claverie and R. Soula, Prog. Polym. Sci., 2003, 28, 619 CrossRef CAS.
- J. Qiu, B. Charleux and K. Matyjaszewski, Prog. Polym. Sci., 2001, 26, 2083 CrossRef CAS.
- M. F. Cunningham, Prog. Polym. Sci., 2008, 33, 365 CrossRef CAS.
- C. Henriquez, C. Bueno, E. A. Lissi and M. V. Encinas, Polymer, 2003, 44, 5559 CrossRef CAS.
- J. Zhang, A. Dong, T. Cao and R. Guo, Eur. Polym. J., 2008, 44, 1071 CrossRef CAS.
- L. Ding, X. F. Jiao, J. P. Deng, W. G. Zhao and W. T. Yang, Macromol. Rapid Commun., 2009, 30, 120 CrossRef CAS.
- W. Saenger, J. Jacob, K. Gessler, T. Steiner, D. Hoffmann, H. Sanbe, K. Koizumi, S. M. Smith and T. Takaha, Chem. Rev., 1998, 98, 1787 CrossRef CAS.
- J. Szejtli, Chem. Rev., 1998, 98, 1743 CrossRef CAS.
- A. Harada, Y. Takashima and H. Yamaguchi, Chem. Soc. Rev., 2009, 38, 875 RSC.
- Y. Zhao, L. Hu, J. F. Stoddart and G. Grüner, Adv. Mater., 2008, 20, 1910 CrossRef CAS.
- J. Zhang and P. X. Ma, Angew. Chem., Int. Ed., 2009, 48, 964 CrossRef CAS.
- M. Guo, M. Jiang, S. Pispas, W. Yu and C. Zhou, Macromolecules, 2008, 41, 9744 CrossRef CAS.
- S. Daoud-Mahammed, P. Couvreur, K. Bouchemal, M. Chéron, G. Lebas, C. Amiel and R. Gref, Biomacromolecules, 2009, 10, 547 CrossRef CAS.
- Y. Liu, C. F. Ke, H. Y. Zhang, J. Cui and F. Ding, J. Am. Chem. Soc., 2008, 130, 600 CrossRef CAS.
- S. Srinivasachari and T. M. Reineke, Biomaterials, 2009, 30, 928 CrossRef.
- H. Ikeda, M. Nakamura, N. Ise, F. Toda and A. Ueno, J. Org. Chem., 1997, 62, 1411 CrossRef CAS.
- F. O'Keeffe, S. A. Shamsi, R. Darcy, P. Schwinte and I. M. Warner, Anal. Chem., 1997, 69, 4773 CrossRef CAS.
- J. Storsberg and H. Ritter, Macromol. Rapid Commun., 2000, 21, 236 CrossRef CAS.
- P. Glöckner, N. Metz and H. Ritter, Macromolecules, 2000, 33, 4288 CrossRef.
- H. Cinar, O. Kretschmann and H. Ritter, Macromolecules, 2005, 38, 5078 CrossRef CAS.
- P. Casper, P. Glöckner and H. Ritter, Macromolecules, 2000, 33, 4361 CrossRef CAS.
- S. Bernhardt, P. Glöckner, A. Theis and H. Ritter, Macromolecules, 2001, 34, 1647 CrossRef CAS.
- J. P. Deng, Q. X. He, Z. L. Wu and W. T. Yang, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2193 CrossRef CAS.
- J. P. Deng, Z. L. Wu, Q. X. He and W. T. Yang, Polym. Adv. Technol., 2008, 19, 1649 CAS.
- T. Uyar, H. S. Gracz, M. Rusa, I. D. Shin, A. El-Shafei and A. E. Tonelli, Polymer, 2006, 47, 6948 CrossRef CAS.
- R. R. Schrock and J. A. Osborn, Inorg. Chem., 1970, 10, 2339 CrossRef.
- Y. L. Loukas, V. Vrak and G. Gregoriadis, Int. J. Pharm., 1996, 144, 225 CrossRef CAS.
- G. Trapani, A. Latrafa and M. Franco, J. Pharm. Sci., 1998, 87, 514 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |