Functional biorenewable polyesters from carvone-derived lactones†
Jennifer R.
Lowe
,
Mark T.
Martello
,
William B.
Tolman
* and
Marc A.
Hillmyer
*
Department of Chemistry and Center for Sustainable Polymers, University of Minnesota, 207 Pleasant St SE, Minneapolis, MN 55455-0431, USA. E-mail: wtolman@umn.edu; hillmyer@umn.edu
First published on 10th December 2010
Abstract
To expand the palette of renewable resource derived monomers that incorporate reactive functionality, the natural product carvone was transformed into two polymerizable lactones, carvomenthide, containing only pendent alkyl groups, and dihydrocarvide, containing an unsaturated moiety. These lactones were polymerized using the catalyst/initiating system diethyl zinc/benzyl alcohol to give aliphatic polyesters with low glass transition temperatures. Good control of the polymer molar masses up to approximately 50 kg mol−1 and products with polydispersity indices below 1.3 were achieved in all cases. Copolymerization of the two lactones was successfully carried out at feed compositions ranging from 3–80 mol% dihydrocarvide, and the ultimate level of dihydrocarvide incorporated into the copolymers was proportional to the feed composition. The pendant double bonds in poly(dihydrocarvide) and copolymers that contain dihydrocarvide were modified by post-polymerization reactions, including epoxidation and radical-induced crosslinking.
Introduction
Petroleum-based monomers have long been the mainstay of the plastics industry, but to reduce dependence on the finite fossil fuel supply there is great interest in developing polymers derived from renewable resources.1 Notable among such renewable starting materials are terpenes and terpenoids,2 commonly occurring natural products derived from various plant sources. Some, such as turpentine (largely pinene) and limonene, have been produced or isolated industrially on very large scales (104 to 105 metric tons annually).3Polymers have been reported from several terpene derivatives including pinene,4limonene,5 and menthol,6 in addition to the most prevalent terpene, isoprene.One terpenoid with the potential to serve as a monomer feedstock is carvone,7 a natural product found in both Mentha spicata (spearmint) and Carum carvi (caraway) oils, that is produced on the scale of 104 metric tons annually.8Hydrogenation of carvone yields carvomenthone and dihydrocarvone (Fig. 1), which are suitable targets for expansion to polymerizable lactones by Baeyer–Villiger oxidation. The oxidation of carvomenthone to carvomenthide (CM) was accomplished in the late nineteenth century,9 but has seldom been explored since, likely because of a lack of practical uses for the resulting carvomenthide.10Dihydrocarvone has been used frequently as a test substrate for selective Baeyer–Villiger oxidations or epoxidations,11 but to our knowledge dihydrocarvide (D) has not been explored as a monomer for aliphatic polyester synthesis.
 | ||
| Fig. 1 Transformation of dihydrocarvone into lactones and their subsequent ring-opening polymerization. | ||
Developing a ‘green’, environmentally friendly Baeyer–Villiger oxidation process for the synthesis of CM and D will be valuable in any effort to make their use practical. The Baeyer–Villiger reaction is one of the most extensively utilized synthetic transformations, with the most common oxidants being organic peracids such as m-chloroperbenzoic acid (m-CPBA) and trifluoroperacetic acid, which are also efficient epoxidation reagents.12 Unfortunately, such organic peracids often are toxic, shock sensitive, require chlorinated solvents, and produce an equivalent of acid that must be removed from the product.13 To alleviate some of the hazards associated with these oxidants, recent work has focused on developing new catalysts for Baeyer–Villiger protocols using hydrogen peroxide and molecular oxygen.13 Additionally, there has been a renewed interest in potassium peroxymonosulfate (KHSO5), the original active oxidant used by Baeyer and Villiger, as a green oxidant.14 Available commercially as a stable potassium triple salt KHSO5·KHSO4·K2SO4 under the trade name Oxone®, it has been shown to convert cyclic ketones to lactones, but yields are often low due to the formation of ω-hydroxy acids.15 In the presence of a ketone under alkali conditions olefins may be efficiently converted to epoxides with Oxone®, thus presenting challenges in efforts to perform selective reactions of substrates with both ketone and olefin functionalities.16 Such challenges must be addressed for the development of the efficient synthesis of D using Oxone®, where lactone formation in the presence of a double bond is required.
Many potential advantages and applications of polymers and copolymers derived from CM and D may be envisioned. The pendent olefin in D could be incorporated unchanged into the product polymer during a metal alkoxide-catalyzed ring-opening polymerization. The resulting polyester would be susceptible to further functionalization, a trait that would be useful for broadening potential applications.17 Examples of such post-polymerization functionalizations include halogenation or epoxidation,18 crosslinking,19 and graft polymer synthesis.20 For example, using the homopolymer polydihydrocarvide (PD) for grafting would produce a bottlebrush type polymer with a graft on every repeat unit.21Copolymerization of D and CM, which contains inert isopropyl rather than isopropenyl groups, would enable control of the number of grafts from each polymer chain or construction of a crosslinked network with a controlled molecular weight between crosslinks. The similarity in chemical structure of the two lactones, especially around the lactone functionality where the ring opening takes place, should favor the formation of a random copolymer. Together, the two renewable resource monomers provide a flexible system that can be tailored to serve as a degradable backbone22 suited to further reactions and modifications for a variety of applications or targets. Similar cyclic ester systems using non-renewable materials have been reported18–20 and have been used to prepare crosslinked nanoparticles for potential biomedical applications.23 Renewable cyclic ester copolymer systems, such as the menthide–lactide system developed previously in our own laboratory,22 can be used for a variety of potential applications but do not have the repeat unit functionality like polymers containing D.
Herein we report procedures for the multi-gram synthesis of CM and D, including a process involving the use of Oxone®. Subsequent homopolymerization of CM to polycarvomenthide (PCM) and D to PD was carried out, and the level of polymerization control and the relative polymerization rates were examined, with a view to drawing comparisons to other ring-opening polymerizations. To demonstrate the potential utility of its pendant double bonds, epoxidation and cross-linking reactions of PD were examined. Finally, copolymers of CM and D were produced with various compositions, allowing the level of functionality of the polymer to be tailored.
Experimental section
General procedures
Toluene (Mallinckrodt, ACS grade) was dried on a home-built solvent column with alumina columns and copper catalyst. ε-Caprolactone (97%, Aldrich) was dried over CaH2 and distilled. Menthide was prepared as previously reported.6 All other chemicals were used as received. Benzene (ACS grade), m-chloroperbenzoic acid (m-CPBA) (77%), Oxone®, diethyl zinc (1.0 M in hexanes), and benzyl alcohol (98%) were acquired from Sigma-Aldrich. (+)-Dihydrocarvone (97%) was acquired from Acros Organics. Wilkinson's catalyst was acquired from Strem Chemicals. Polymerizations were set-up in a nitrogen-atmosphere dry box and were carried out under a nitrogen blanket.Characterization
Size Exclusion Chromatography was carried out on an Agilent 1000 system with a Varian PLgel 5 guard column and three Varian PLgel Mixed C columns using a Hewlett-Packard 1047A refractive index detector. CHCl3 was used as a solvent and narrow molecular weigh distribution polystyrene standards were used for calibration. 1H NMR Spectroscopy was performed on a Varian Inova 500 MHz instrument (125 MHz for 13C). Small molecule 1H NMR experiments were performed with a minimum of 32 transients. Polymer 1H NMR experiments were performed with a delay time of at least 10 s and a minimum of 16 transients (13C NMR: 2.5 s delay time and a minimum of 64 transients). Differential Scanning Calorimetry was carried out on a Texas Instrument QA1000 DSC using an indium standard. The hermetically sealed samples were cooled to −80 °C, heated at 10 °C min−1 to at least 30 °C, allowed to equilibrate, cooled at 10 °C min−1 to −80 °C, allowed to equilibrate, and heated a final time at 10 °C min−1 to at least 30 °C. The glass transition temperatures were determined from the second heating cycle. GC-MS experiments were carried out on an Agilent Technologies 7890A gas chromatograph and an Agilent Technologies 5975C VL mass spectrometer detector using electron ionization.Synthesis of carvomenthide
A Pressure Products Industries, Inc. high-pressure hydrogenation unit was charged with Wilkinson's catalyst (1.48 g, 1.6 mmol) and placed under reduced pressure (25 °C, 5 Torr). After purging with argon, the hydrogenation unit was then charged with (+)-dihydrocarvone (26.1 mL, 159 mmol), toluene (125 mL), and hydrogen (280 psig). The reaction was allowed to proceed at room temperature for 42 h. The reaction mixture was then vacuum filtered using Millipore 0.22 µm paper. The toluene was removed using rotary evaporation and the carvomenthone product was purified using vacuum distillation (boiling point 40 °C at 175 mTorr) to give 18.45 g (120 mmol, 75.1% yield) of a colorless, transparent oil. 1H NMR (500 MHz, CDCl3): 2.25–2.45 (m, 2H major, 3H minor), 1.98–2.1 (m, 2H major), 1.79–1.88 (m, 1H major, 1H minor), 1.35–1.75 (m, 3H major, 5H minor), 1.26 (qd, J = 3, 12.5 Hz, 1H major), 1.06 (d, J = 7 Hz, 3H minor), 0.98 (d, J = 7 Hz, 3H major), 0.82–0.90 (m, 6H major, 6H minor). 13C NMR (125 MHz, CDCl3): 213.7, 46.7, 45.5, 45.0, 35.2, 32.9, 29.0, 19.7, 19.4, 14.5 (major isomer), 214.6, 44.9, 44.4, 43.0, 31.5, 30.7, 25.0, 20.14, 20.06, 16.0 (minor isomer). Mass spectrum (70 eV) m/z (relative intensity): 154 (26, M+), 112 (10), 111 (100), 110 (10), 97 (15), 95 (17), 83 (19), 69 (23), 56 (10), 55 (74).A 250 mL round bottom flask was charged with carvomenthone (12.67 g, 82.2 mmol), methylene chloride (158 mL), and m-chloroperbenzoic acid (25.37 g, 113 mmol). The reaction flask was capped and the mixture was allowed to stir for approximately 2 days. The reaction mixture was filtered to remove solids and was then washed with sodium bisulfite solution (5 g in 100 mL), saturated sodium bicarbonate solution (3 × 100 mL), and sodium chloride brine (2 × 100 mL). The organic layer was dried over MgSO4 and filtered. The solvent was removed by rotary evaporation to give a pale yellow oil which was then vacuum distilled (boiling point 75 °C at 50 mTorr) to give a colorless, transparent oil (12.69 g, 90.7%). 1H NMR (500 MHz, CDCl3): 4.4 (qdd, J = 6.5, 9.5, 13.5 Hz, 1H), 2.4 (m, 2H), 1.84 (m, 1H), 1.73 (m, 1H), 1.59 (m, 1H), 1.54 (m, 1H), 1.45 (m, 1H), 1.40 (m, 1H), 1.26 (d, J = 6.5 Hz, 3H), 0.79–0.81 (two closely spaced doublets, J = 7 Hz, 6H). 13C NMR (125 MHz, CDCl3): major isomer 175.7 (C), 76.6 (CH), 40.3 (CH), 38.1 (CH2), 35.8 (CH2), 33.6 (CH), 31.3 (CH2), 22.6 (CH3), 18.8 (CH3), 18.5 (CH3); minor isomer 174.3 (C), 76.4 (CH), 38.8 (CH), 38.4 (CH2), 32.2 (CH2), 30.1 (CH2), 29.0 (CH), 22.3 (CH3), 20.7 (CH3), 20.4 (CH3). Mass spectrum (70 eV) m/z (relative intensity): 170 (0.1, M+), 127 (20), 126 (72), 97 (10), 84 (22), 83 (100), 82 (10), 70 (33), 69 (36), 67 (11), 57 (10), 56 (34), 55 (78) (Fig. S1, S4, and S6†).
Synthesis of dihydrocarvide
In a 1 L round bottom flask, dihydrocarvone (11.1 mL, 10.3 g, 67.8 mmol) was dispersed in deionized water (200 mL) and methanol (200 mL). Portions consisting of ∼7.4 g (24.1 mmol KHSO5) of Oxone® and ∼5.3 g (63 mmol) sodium bicarbonate were added at reaction times of 0 h, 4 h, 21 h, and 28 h. After 48 h, the reaction mixture was filtered to remove solids and the methanol was removed using rotary evaporation. The remaining aqueous phase was extracted with portions of diethyl ether (3 × 60 mL). The diethyl ether layers were collected and washed sequentially with portions of sodium bisulfite solution (1 × 4 g in 70 mL) and DI water (2 × 70 mL). The organic layer was dried over MgSO4 and concentrated to give a pale yellow oil. GC-MS and 1H NMR spectroscopy analysis showed approximately 86% conversion to the desired lactone and 10% to the epoxide side product. A silica gel column with a 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 solvent mixture of hexanes and ethyl acetate was used to remove the remaining dihydrocarvone starting material, which elutes first. A second column with the same conditions was used to separate the lactone from the epoxide. The trans isomer of the lactone elutes first, followed by the stereoisomers of the epoxide, and finally the second (cis) isomer of the lactone (Rf = 0.45 for the translactone, 0.39 and 0.36 for the epoxide isomers, and 0.35 for the cislactone). The dihydrocarvide was vacuum distilled (oil bath at 65 °C at a pressure of 100 mTorr) to give a colorless, transparent oil (4.78 g, 42%). 1H NMR (500 MHz, CDCl3): 4.76 (s, 1H), 4.73 (s, 1H), 4.47 (ddq, J = 6.5, 13, 16 Hz, 1H), 2.55–2.75 (m, 2H), 2.3 (m, 1H), 1.9–2.0 (m, 2H), 1.73 (s, 3H), 1.55–1.70 (m, 2H), 1.37 (d, J = 6.5 Hz, 3H). 13C NMR (125 MHz, CDCl3): 174.9 (C), 148.6 (C), 110.4 (CH2), 76.8 (CH), 42.0 (CH), 40.4 (CH2), 36.0 (CH2), 34.5 (CH2), 22.8 (CH3), 20.4 (CH3). Gas chromatograph: retention time, percent area (compound identity): 13.73 min 0.5% (epoxide isomer), 13.86 min 0.3% (epoxide isomer), 14.44 min 95.1% (dihydrocarvide isomer), 14.51 min 4.1% (dihydrocarvide isomer). Mass spectrum (70 eV) of 14.4 min in GC (dihydrocarvide) m/z (intensity): 168 (6, M+), 126 (13), 125 (28), 111 (24), 110 (12), 109 (17), 108 (35), 107 (12), 97 (10), 96 (22), 95 (14), 93 (21), 83 (10), 82 (25), 81 (31), 79 (15), 71 (11), 69 (21), 68 (91), 67 (100), 55 (34), 54 (10), 53 (26) (Fig. S7, S9, and S10†).
:1 solvent mixture of hexanes and ethyl acetate was used to remove the remaining dihydrocarvone starting material, which elutes first. A second column with the same conditions was used to separate the lactone from the epoxide. The trans isomer of the lactone elutes first, followed by the stereoisomers of the epoxide, and finally the second (cis) isomer of the lactone (Rf = 0.45 for the translactone, 0.39 and 0.36 for the epoxide isomers, and 0.35 for the cislactone). The dihydrocarvide was vacuum distilled (oil bath at 65 °C at a pressure of 100 mTorr) to give a colorless, transparent oil (4.78 g, 42%). 1H NMR (500 MHz, CDCl3): 4.76 (s, 1H), 4.73 (s, 1H), 4.47 (ddq, J = 6.5, 13, 16 Hz, 1H), 2.55–2.75 (m, 2H), 2.3 (m, 1H), 1.9–2.0 (m, 2H), 1.73 (s, 3H), 1.55–1.70 (m, 2H), 1.37 (d, J = 6.5 Hz, 3H). 13C NMR (125 MHz, CDCl3): 174.9 (C), 148.6 (C), 110.4 (CH2), 76.8 (CH), 42.0 (CH), 40.4 (CH2), 36.0 (CH2), 34.5 (CH2), 22.8 (CH3), 20.4 (CH3). Gas chromatograph: retention time, percent area (compound identity): 13.73 min 0.5% (epoxide isomer), 13.86 min 0.3% (epoxide isomer), 14.44 min 95.1% (dihydrocarvide isomer), 14.51 min 4.1% (dihydrocarvide isomer). Mass spectrum (70 eV) of 14.4 min in GC (dihydrocarvide) m/z (intensity): 168 (6, M+), 126 (13), 125 (28), 111 (24), 110 (12), 109 (17), 108 (35), 107 (12), 97 (10), 96 (22), 95 (14), 93 (21), 83 (10), 82 (25), 81 (31), 79 (15), 71 (11), 69 (21), 68 (91), 67 (100), 55 (34), 54 (10), 53 (26) (Fig. S7, S9, and S10†).
Homopolymerization of carvomenthide
In a representative reaction, a 15 mL pressure vessel was charged with CM (0.198 g, 1.16 mmol), benzyl alcohol (1.20 µL, 1.16 × 10−2 mmol), and ZnEt2 (36 µL, 3.6 × 10−2 mmol) under a nitrogen atmosphere. The vessel was capped and placed in a 100 °C oil bath to stir. After 8 h, the vessel was removed from the oil bath and allowed to cool. Three drops of 0.15 M HCl were added to quench the reaction and 0.75 mL of CH2Cl2 was used to dissolve the polymer. The polymer was precipitated in approximately 15 mL of chilled (∼8 °C) methanol. The methanol was decanted and the polymer dried under vacuum to give 0.178 g (90% yield) of colorless high viscosity material. 1H NMR: 4.84 (m, 1H), 2.22 (dd, J = 6, 15 Hz, 1H), 2.07 (dd, J = 7, 15 Hz, 1H), 1.62–1.8 (m, 2H), 1.54 (m, 1H), 1.44 (m, 1H), 1.29 (m, 1H), 1.21 (m, 1H), 1.17 (d, J = 6.5 Hz, 3H), 0.82 (2d, J = 7 Hz, 6H). 13C NMR: 173.6 (C), 71.0 (CH), 40.7 (CH), 36.4 (CH2), 33.5 (CH2), 29.9 (CH), 26.7 (CH2), 20.0 (CH3), 19.2 (CH3), 18.7 (CH3).Homopolymerization of dihydrocarvide
In a representative reaction, a catalyst solution containing 12.6 µL benzyl alcohol and 244 µL of 1.0 M ZnEt2 in hexanes was prepared. A 7.4 mL vial with a PTFE-lined cap was charged with D (49.6 mg, 0.30 mmol) and 5.3 µL of catalyst solution (2.5 × 10−3 mmol benzyl alcohol, 5.1 × 10−3 mmol ZnEt2) under a nitrogen atmosphere. The vial was capped and placed in a 100 °C oil bath to stir for 8 h. The reaction was quenched by opening the vial to air. The polymer was characterized by SEC, 1H NMR, and 13C NMR spectroscopy. 1H NMR (500 MHz, CDCl3): 4.84 (m, 1H), 4.76 (s, 1H), 4.72 (s, 1H), 2.52 (m, 1H), 2.30 (m, 2H), 1.63 (s, 3H), 1.5–1.2 (m, 4H), 1.16 (d, J = 6 Hz, 3H). 13C NMR (125 MHz, CDCl3): 172.2 (C), 145.7 (C), 112.8 (CH2), 71.0 (CH), 44.0 (CH2), 39.5 (CH), 33.7 (CH2), 28.8 (CH2), 20.1 (CH3), 18.6 (CH3).Relative rate studies of the polymerization of carvomenthide, dihydrocarvide, menthide, and ε-caprolactone
In an inert atmosphere dry box, CM (998.1 mg, 5.94 mmol), benzyl alcohol (3.7 µL, 3.6 × 10−2 mmol), and ZnEt2 (83.0 µL, 8.3 × 10−2 mmol) were stirred in a scintillation vial for approximately 5 min. Approximately 70 µL aliquots of the reaction mixture were injected into a series of 3.7 mL vials equipped with stir bars and PTFE-line caps. The vials were placed into a preheated 100 °C oil bath and the polymerizations were allowed to proceed for varying lengths of time. To quench a reaction, a vial was removed from the oil bath, cooled to <50 °C, opened to air, and quenched with 4 µL of 0.15 M HCl in water. 1H NMR spectroscopy and SEC in CHCl3 were used to characterize the samples. Similar experiments were carried out using ε-caprolactone, menthide, and D.Copolymerization of carvomenthide and dihydrocarvide
In a typical reaction, a 3.7 mL vial was charged with a stir bar, CM (75.1 mg, 0.441 mmol), D (73.7 mg, 0.439 mmol), benzyl alcohol (0.93 µL, 9.0 × 10−3 mmol), and ZnEt2 (18.5 µL, 18.5 × 10−3 mmol). The vial was capped and moved to a 100 °C oil bath for 8 h. The vial was removed from the oil bath, cooled to <50 °C, opened to air, and quenched with 0.15 M HCl (10 µL). Samples were dissolved in ∼0.25 mL of CH2Cl2 and precipitated into 8 mL of hexane chilled in dry ice. The resulting polymers were analyzed using 1H NMR spectroscopy and SEC. Some samples were further characterized using DSC (Fig. S12–S14†).Epoxidation of poly(dihydrocarvide-co-carvomenthide)
In a 3.7 mL vial, poly(dihydrocarvide-co-carvomenthide) containing 10 mol% D (49.5 mg, 17 kg mol−1) was dissolved in CDCl3 (0.82 mL). m-CPBA (13.7 mg) was added and the reaction mixture was allowed to stir at room temperature. After 24 h, 1H NMR spectroscopy showed 100% conversion. The polymer was characterized by SEC, DSC, and 1H NMR spectroscopy. 1H NMR (500 MHz, CDCl3): 4.84 (m, 100H), 2.43–2.66 (m, 30H), 2.02–2.30 (m, 200H), 1.38–1.80 (m, 420H), 1.08–1.35 (m, 490H), 0.74–0.91 (m, 630H).Crosslinking of polydihydrocarvide
In a representative reaction, polydihydrocarvide (polyD) (48.9 mg, 10 kg mol−1), AIBN (2.3 mg), and 3,6-dioxane-1,8-octanedithiol (2.40 µL) were dissolved in benzene (0.1 mL) in a 3.7 mL vial. The vial was capped and heated in an 80 °C oil bath for 5 h. After the reaction, the vial was cooled and the solid contents were washed with portions of DI water (3 × 1 mL). The solid polymer was dried under vacuum at 100 mTorr and 60 °C for 48 hours to remove the excess thiol and the benzene solvent. After drying, no trace of sulfur odor was detected. The crosslinked polymers were characterized by solvent extraction to determine the gel fraction. To determine the gel fraction, 50 mg samples of polymer were placed into 1.8 mL of CHCl3 for 48 h. After 48 h, the excess solvent was decanted off and the swollen polymer samples were weighed to determine the solvent absorption. The samples were then dried under vacuum for 24 h and weighed to determine the gel fraction.Results and discussion
Syntheses of carvomenthide (CM) and dihydrocarvide (D)
Dihydrocarvone was hydrogenated using a modified literature procedure24 to produce carvomenthone on an 18 g scale (Fig. 1). The carvomenthone was then oxidized using 1.5 equivalents of m-CPBA to produce CM on a 15 g scale. After distillation, 1H NMR and 13C NMR spectroscopy and GC-MS spectrometry were used to confirm the identity of the carvomenthide product through comparison to literature spectra24 (see Experimental section and Fig. S1–S6†). Only the expected Baeyer–Villiger oxidation product derived from migration of the more substituted carbon14 was formed, with no apparent insertion of the oxygen between the carbonyl and the methylene 6 position.To prepare D, a method using Oxone® was developed to perform the Baeyer–Villiger oxidation of the cyclic ketone with minimal epoxidation of the alkene (see the Experimental section). The preliminary results reported here are promising for the development of a ‘green’ Baeyer–Villiger protocol in the presence of alkenes using Oxone®. Analysis of the crude product showed 86% conversion of the ketone to the desired Baeyer–Villiger product (D) with only 10% conversion to the epoxide (4% of unreacted starting material remained). The competition between the two oxidation pathways, giving rise to the observed product distributions, appears consistent with those reported for directed epoxidation studies (it should be noted that the co-solvent was acetonitrile for these epoxidation studies).16 Notably, significant ring-opening of the lactone by either water or methanol was not observed under these conditions.15 After distillation, 1–2% of the epoxide impurity was identified by 1H NMR spectroscopy. Separation of the epoxide from the unsaturated lactone proved challenging and attempts to remove the epoxide entirely led to lower yields as multiple columns were necessary and much of the minor stereoisomer (∼20% of the mixture, in keeping with the isomer ratio in the dihydrocarvone starting material) was lost. The mixture of D stereoisomers obtained (Fig. 1) was 4:1 trans:cis, as in the starting material. This conclusion is based on the results of GC-MS analysis (two compounds with nearly identical retention times, roughly 4:1 integral areas, and identical mass spectra). In addition, we were able to isolate a small amount of the pure trans isomer in low yield, and it was identified by comparison of 1H and 13C NMR spectroscopy and GC data to information reported in the literature10b (see Fig. S7–S10†).
Homopolymerizations of CM and D
Homopolymerization of CM was carried out in the bulk using diethyl zinc as a catalyst and benzyl alcohol as an initiator at a variety of monomer to initiator ratios (Table 1). 1H NMR spectroscopy was used to analyze the PCM molar masses and draw comparisons to data for the related homopolymers polymenthide (PM) and PD (Fig. 2). The integration value of the resonance due to the methine proton in the polymer repeat unit at 4.84 ppm was compared to that for the methylene protons in the benzyl group α chain end (5.1 ppm, not shown) to determine Mn, assuming one benzyl group per polymer chain. The Mn values calculated using these integral ratios corresponded well with Mn values determined by SEC relative to polystyrene standards (Table 1). A comparison of the experimental monomer to initiator ratios to the Mn of the resulting polymers indicates that good control of the molecular weight is possible up to about 50 kg mol−1; above that, there is a little increase in the molar mass. This upper limit on Mn has been previously reported in lactone polymerizations and it has been suggested that a mixture of trace impurities in the monomer feedstock or catalyst is responsible for adventitious initiation.25| Monomer | [M]:[C]:[I] |
M n Calc.a/kg mol−1 | M n 1H NMR/kg mol−1 | M n SEC/kg mol−1 | PDI |
|---|---|---|---|---|---|
| a Calculated molecular weight is based upon the monomer to initiator ratio and the conversion by 1H NMR spectroscopy. | |||||
| CM | 5.9:2:1 |
0.94 | 0.75 | 1.5 | 1.17 |
| CM | 30:2:1 |
5.1 | 4.1 | 5.6 | 1.10 |
| CM | 62:2:1 |
10.5 | 9.1 | 10.4 | 1.10 |
| CM | 100:2:1 |
13.9 | 11.8 | 14.1 | 1.08 |
| CM | 165:2:1 |
27.4 | 23 | 25.5 | 1.08 |
| CM | 319:2:1 |
53.1 | 31 | 53.4 | 1.12 |
| CM | 670:2:1 |
111 | 43 | 62.3 | 1.16 |
| D | 6.8:2:1 |
1.2 | 1.3 | 0.7 | 1.26 |
| D | 34:2:1 |
5.6 | 3.9 | 1.8 | 1.21 |
| D | 77:2:1 |
12.7 | 9.7 | 2.9 | 1.31 |
| D | 118:2:1 |
19.5 | 13 | 5.5 | 1.24 |
| D | 226:2:1 |
37.2 | 21 | 6.9 | 1.30 |
| D | 347:2:1 |
57.9 | 26 | 7.8 | 1.30 |
| D | 564:2:1 |
93.7 | 59 | 10.5 | 1.24 |
| D | 30:2:0 |
7.1 | 1.42 |
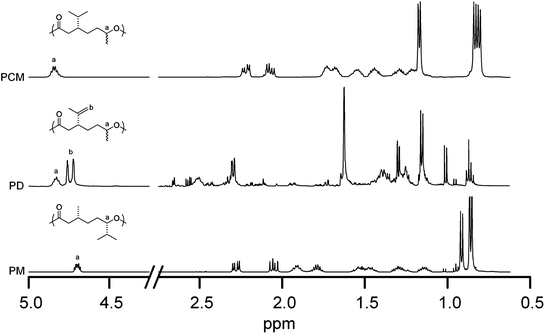 | ||
| Fig. 2 1H NMR spectra of polycarvomenthide (top, PCM), polydihydrocarvide (middle, PD), and polymenthide (bottom, PM). | ||
Homopolymerization of D to PD was also carried out in the bulk using diethyl zinc and benzyl alcohol (Table 1). 1H NMR spectroscopy (Fig. 2) was used to analyze the molar mass of PD in the same manner as for PCM. Due to the structural similarities of the two repeat units, the methine proton peak adjacent to the heteroatom oxygen occurs at 4.84 ppm in both PD and PCM. The polymerization of D leaves the olefinic protons, present at 4.76 and 4.72 ppm in the PDspectrum, unmodified. Comparing the integration values of the olefinic peaks to the 4.84 ppm methine peak confirms that the olefin survives the polymerization intact and is thus available for further reactions.
The Mn values for PD (Table 1) show that there is a disparity between the measured and calculated Mn values even at low molar masses. In attempts to understand this issue, a control polymerization was performed under conditions similar to those used in the polymerization with a 34:2:1 monomer to catalyst to initiator ratio, but with no initiator present. After 4 h of reaction time using a 30:2 monomer to catalyst ratio, the polymerization had a conversion of 60% with an SEC Mn of 7.1 kg mol−1 and a PDI of 1.42. The fact that polymer is generated in the absence of added benzyl alcohol indicates that there is fortuitous initiator present in the dihydrocarvide monomer D. We speculate that this could be from some of the residual epoxide (about a 1 mol% impurity in CM by GC) which, if it underwent ring-opening at the increased temperature of the polymerizations, could serve as an initiator for ring-opening polymerization of the dihydrocarvide. Of course, other low levels of an unidentified impurity could be responsible for initiation. Thus, the ability of the dihydrocarvide monomer mixture to polymerize without additional initiator creates some difficulty in achieving higher molecular weights in a controlled manner at the CM purity levels utilized here.
That the 1H NMR spectroscopy calculated values and the SEC values of Mn for PCM are nearly identical (up to about 50 kg mol−1) suggests that the hydrodynamic radii of the polystyrene standards (used to calibrate the SEC) and PCM are nearly equivalent at comparable molar masses. The only difference in structure between PD and PCM is the unsaturation in one pendant group of every 7 backbone atoms (Fig. 1), and thus we expect little difference in hydrodynamic volumes between PCM and PD. Thus we attribute the large difference in the 1H NMR spectroscopy and SEC Mn values for PD (Table 1) to unaccounted endgroups in the NMR spectra stemming from fortuitous initiator in D (estimated to be 1.4 mol%)26 and not due to hydrodynamic volume differences between PD and polystyrene.
Comparison of the 1H NMR spectra of the structurally similar PCM, PD, and PM reveals some key differences that allow them to be readily distinguished (Fig. 2). As mentioned, the methine proton on the carbon adjacent to the backbone oxygen has identical shifts of 4.84 ppm for PCM and PD owing to the presence of a methyl group on the same carbon in both cases. The replacement of that methyl group with an isopropyl group in PM leads to a slight shift of that peak to 4.73 ppm.16 Further differences may be seen in the position of the methyl group resonances in the 1H NMR spectra. Notwithstanding these different spectroscopic properties, the structural congruencies for the three homopolymers result in similar glass transition temperatures: −27 °C for PCM, −26 °C for PM, and −20 °C for PD. The lack of crystallinity in PCM and PD can be attributed both to mixed configuration of the methyl substituent and the presence of the bulky isopropyl substituent, as in PM (which is stereoregular).
To assess the level of control in the homopolymerizations of CM and D, we explored the change in PDI and Mn as a function of conversion (Fig. 3) for samples produced using a 175:2:1 monomer to catalyst to initiator ratio. ε-Caprolactone (CL) was polymerized under identical conditions for comparison. A linear increase in Mn with conversion was observed for all three monomers (R2 values for the linear regressions were 0.98 or greater). For CL and CM, these regressions had intercepts of approximately the monomer molecular weight. A fit of the Mnversus conversions data for D gave a smaller slope than for CM consistent with the presence of fortuitous initiator as described above. The plot of PDI as a function of conversion (Fig. 3) illustrates the nature of the polymerizations of CM and D. In the case of CM, the PDI remained below 1.15, even at high conversions (greater than 95%). In the case of D, the PDI remained steady between 1.2 and 1.3 at conversions up to 88%. In contrast, CL showed a large increase in PDI at higher conversions, with the PDI approaching 2.0 as the conversion approaches 99%, likely due to transesterification reactions that PCL is known to undergo.27 The fact that the PDI remained low for both D and CM indicates that polymerizations can be carried out to high conversions without changing the distribution of the chain sizes. Additionally, the lack of apparent transesterification indicates that D and CM could be used for the formation of discrete block copolymers because they would be less likely to undergo chain transfer during the ring-opening polymerization. Similar results were found previously for menthide (M), which can be used for the preparation of block copolymers with polylactide.22
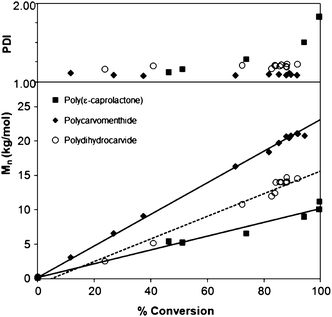 | ||
| Fig. 3
Polymer molar mass (Mn, determined from SECvs.polystyrene standards) and polydispersity (PDI) as a function of % conversion. Polymerizations were carried out in the bulk at 100 °C using 175:2:1 monomer to diethyl zinc to benzyl alcohol ratio. | ||
Copolymerization of CM and D
Polymers with different ratios of CM and D were targeted in order to control the level of olefin functionality on the polymer chain. Specifically, copolymers of CM and D were produced with CM content of 2.7 to 80 mol% by polymerizing a mixture of the two monomers using conditions identical to those used in the homopolymerization reactions (Table 2). 1H NMR spectroscopy was used to analyze the comonomer content in the resulting polymers (Fig. S12†). In both homopolymers, the methine proton of the repeat unit appears at 4.84 ppm (Fig. 2). The relative amounts of the two monomers were determined by comparing the area of this methine proton peak, which contains one proton from every repeat unit be it CM or D, with the area of the peak due to the two alkene protons in the Drepeat units (4.75 ppm, Fig. 2). In all cases, the incorporation level of CM and D reflected the composition of the original feed. In all cases, the SEC traces were monomodal (Fig. S13†) with PDI values between 1.1 and 1.2, lending support to the fact that copolymers rather than separate homopolymers were produced. Analysis of select samples by DSC showed a single Tg.| % D (feed) | % D (by 1H NMR) | T g °C (by DSC) |
|---|---|---|
| a All copolymerization conversions were >95%. The copolymerizations were performed in the absence of solvent and were analyzed directly from the reaction mixture. | ||
| 0 | 0 | −27 |
| 2.7 | 3.9 | −28 |
| 5.1 | 7.2 | |
| 11 | 13 | |
| 21 | 24 | −26 |
| 32 | 34 | |
| 41 | 44 | |
| 50 | 53 | −22 |
| 60 | 60 | |
| 71 | 72 | |
| 80 | 80 | |
| 100 | 100 | −20 |
Post-polymerization reactions of PD
A variety of post-polymerization reactions are possible with PD and related copolymers including epoxidation, halogenations, addition of a hydroxyl group, or crosslinking by a variety of methods.17 In initial studies we focused on epoxidation and crosslinking reactions. Epoxidation of a copolymer containing 10 mol% D and the balance CM was carried out using 1.5 equivalents of m-CPBA. After 24 h, 1H NMR analysis showed the disappearance of the proton resonances at 4.72 ppm associated with the alkene protons of the pendent double bonds and the appearance of peaks corresponding to an unopened epoxide ring between 2.5 and 2.7 ppm. SEC analysis showed a drop in molecular weight (14 kg mol−1 to 4 kg mol−1) with an increase in PDI to 1.65. This is consistent with some degradation during the reaction, likely due to the acidic conditions. Homopolymer made directly from an epoxylactone monomer and thus possessing the same repeat unit structure as the epoxidized polymer reported here had only low Mn values by SEC, but in the previous work, there was some branching which would not be the case here.7 It is possible that buffering the reaction or running the reaction for a shorter period of time would result in less acid-induced degradation of the polymer chains if epoxidized polymer is desired.Crosslinking of PD was carried out using a dithiol with a small amount of radical initiator to join the pendant double bonds. Different ratios of crosslinkers were used resulting in different gel fractions and molecular weights between crosslinks, as shown in Table 3. At 0.25 and 0.13 equivalents of dithiol, crosslinked networks capable of absorbing solvent without dissolving were formed. When only 0.05 equivalents of dithiol were used, however, there was almost no gel fraction. Little crosslinking was achieved with the poly(D-co-CM) copolymer containing 10% D, but it is possible that repeated additions of radical initiator are needed to allow further crosslinking due to the lower concentration of double bonds and the increased reaction time needed. It is also possible that primary cyclization, or crosslinking back onto the same chain, occurs, a precedented phenomenon that is especially likely when the concentration of crosslinkable groups is low.28
Conclusions
The natural product dihydrocarvone was converted into the seven-membered lactones carvomenthide and dihydrocarvide on a multi-gram scale. Both monomers were effectively converted to the corresponding aliphatic polyesters with low PDI values in bulk polymerizations using diethyl zinc as the catalyst and benzyl alcohol as the initiator. The polymers of both lactones exhibited low glass transition temperatures (less than −20 °C). When copolymerized, carvomenthide and dihydrocarvide form polymers with controllable comonomer content. The reactive alkene side groups were epoxidised and also used to crosslink these materials. Further post-polymerization functionalization or copolymerization strategies could be undertaken in the future as these renewable materials represent a versatile system.Acknowledgements
The authors would like to acknowledge the financial support provided by the NSF (CHE-0842654) and the Center for Sustainable Polymers at the University of Minnesota. We thank Tyler Stack for contributions to the study of dihydrocarvone oxidation selectivity.Notes and references
- (a) J. B. van Beilen and Y. Poirier, Plant J., 2008, 54, 684–701 CrossRef CAS; (b) L. Yu, K. Dean and L. Li, Prog. Polym. Sci., 2006, 31, 576–602 CrossRef CAS; (c) M. S. Lindblad, Y. Liu, A. C. Albertsson, E. Ranucci and S. Karlsson, Adv. Polym. Sci., 2000, 157, 139–161.
- A. J. D. Silvestre and A. Gandini, in Monomers, Polymers and Composites from Renewable Resources, ed. M. N. Belgacem and A. Gandini, Elsevier, Amsterdam, 2009, pp. 17–38 Search PubMed.
- (a) Limonene: A. F. Filipsson, J. Bard and S. Karlsson, Concise International Chemical Assessment Document No. 5: Limonene, World Health Organization, International Programme on Chemical Safety, Geneva, 1998 Search PubMed; (b) Turpentine: J. J. W. Coppen and G. A. Hone, Non-Wood Forest Products, Natural Resources Institute, Food and Agricultural Organization of the United Nations, Rome, 1995, ch. 1 Search PubMed.
- W. J. Roberts and A. R. Day, J. Am. Chem. Soc., 1950, 72, 1226–1230 CrossRef CAS.
- (a) C. M. Byrne, S. D. Allen, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2004, 126, 11404–11405 CrossRef CAS; (b) K. Xu, C. Mingcai, K. Zhang and J. Hu, Polymer, 2004, 45, 1133–1140 CrossRef CAS; (c) M. Modena, R. B. Bates and C. S. Marvel, J. Polym. Sci., Part A: Gen. Pap., 1965, 3, 949–960 CrossRef CAS.
- D. Zhang, M. A. Hillmyer and W. B. Tolman, Biomacromolecules, 2005, 6, 2091–2095 CrossRef CAS.
- J. R. Lowe, W. B. Tolman and M. A. Hillmyer, Biomacromolecules, 2009, 10, 2003–2008 CrossRef CAS.
- (a) C. C. C. R. De Carvalho and M. M. R. Da Fonseca, Food Chem., 2006, 95, 413–422 CrossRef CAS; (b) A. F. Filipsson, J. Bard and S. Karlsson, Concise International Chemical Assessment Document No. 5: Limonene, World Health Organization, International Programme on Chemical Safety, Geneva, 1998 Search PubMed.
- A. Baeyer and V. Villiger, Ber. Dtsch. Chem. Ges., 1899, 32, 3625–3633 CrossRef.
- (a) V. Alphand and R. Furstoss, Tetrahedron: Asymmetry, 1992, 3, 379–382 CrossRef; (b) P. Cernuchova and M. D. Mihovilovic, Org. Biomol. Chem., 2007, 5, 1715–1719 RSC; (c) D. V. Rial, P. Cernuchova, J. B. van Beilen and M. D. Mihovilovic, J. Mol. Catal. B: Enzym., 2008, 50, 61–68 CrossRef CAS.
- (a) O. Abril, C. C. Ryerson, C. Walsh and G. M. Whitesides, Bioorg. Chem., 1989, 17, 41–52 CrossRef CAS; (b) S. Meziane, P. Lanteri, R. Longeray and C. Arnaud, C. R. Acad. Sci., Ser. IIc: Chim., 1998, 1, 91–94 CrossRef CAS; (c) A. Corma, M. T. Navarro, L. Nemeth and M. Renz, Chem. Commun., 2001, 2190–2191 RSC; (d) M. Renz, R. Blasco, A. Corma, V. Fornes, R. Jensen and L. Nemeth, Chem.–Eur. J., 2002, 8, 4708–4717 CrossRef CAS; (e) A. Brunetta and G. Strukul, Eur. J. Inorg. Chem., 2004, 1030–1038 CrossRef CAS.
- G. R. Krow, Org. React., 1993, 43, 251 CAS.
- G.-J. ten Brink, I. W. C. E. Arends and R. A. Sheldon, Chem. Rev., 2004, 104, 4105–4123 CrossRef.
- M. Renz and B. Meunier, Eur. J. Org. Chem., 1999, 737–750 CrossRef CAS.
- R. J. Kennedy and A. M. Stock, J. Org. Chem., 1960, 25, 1901–1906 CrossRef CAS.
- (a) Z. Wang, Y. Tu, M. Frohn and Y. Shi, J. Org. Chem., 1997, 62, 2328–2329 CrossRef CAS; (b) Z. Wang, Y. Tu, M. Frohn, J. Zhang and Y. Shi, J. Am. Chem. Soc., 1997, 119, 11224–11235 CrossRef CAS.
- P. Lecomte, R. Riva, S. Schmeits, J. Rieger, K. Van Busele, C. Jerome and R. Jerome, Macromol. Symp., 2006, 240, 157–165 CrossRef CAS.
- D. Mecerreyes, R. D. Miller, J. L. Hedrick, C. Detrembleur and R. Jerome, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 870–875 CrossRef CAS.
- A.e. Van der Ende, E. J. Kravitz and E. Harth, J. Am. Chem. Soc., 2008, 130, 8706–8713 CrossRef CAS.
- B. Parrish and T. Emrick, Macromolecules, 2004, 37, 5863–5865 CrossRef CAS.
- S. J. Lord, S. S. Sheiko, I. LaRue, H. Lee and K. Matyjaszewski, Macromolecules, 2004, 37, 4235–4240 CrossRef CAS.
- (a) C. L. Wanamaker, L. E. O'Leary, N. A. Lynd, M. A. Hillmyer and W. B. Tolman, Biomacromolecules, 2007, 8, 3634–3640 CrossRef CAS; (b) C. L. Wanamaker, W. B. Tolman and M. A. Hillmyer, Biomacromolecules, 2009, 10, 443–448 CrossRef CAS.
- For a review of the subject, see: R. J. Pounder and A. P. Dove, Polym. Chem., 2010, 1, 260–271 Search PubMed.
- A. Furstner and P. Hannen, Chem.–Eur. J., 2006, 12, 3006–3019 CrossRef.
- (a) D. R. Witzke and R. Narayan, Macromolecules, 1997, 30, 7075–7085 CrossRef CAS; (b) H. R. Kricheldorf, Eur. Polym. J., 1989, 25, 585–591 CrossRef CAS; (c) A. J. Nijenhuis, D. W. Grijpma and A. J. Pennings, Macromolecules, 1992, 25, 6419–6424 CrossRef CAS.
- Assuming the SEC Mn for the Dpolymerization without benzyl alcohol is accurate, then the level of fortuitous initiator would be 1.4 mol% [i.e., a [D]0/[fortuitous initiator]0 = 70 would give a PDMn = 7.1 kg mol−1 at 60% conversion]. Consider the polymerization of D at 564:2:1 monomer to catalyst to initiator ratio in Table 1. The conversion of this polymerization was 99% and the level of benzyl alcohol utilized was 0.18 mol%. If the fortuitous initiator level was also 1.4 mol% in this polymerization, the expected amount of total initiator would be 1.58 mol% and thus at 99% conversion the expected Mn would be (1/0.0158) × 168 g mol−1 × 0.99 = 10.5 kg mol−1. This value is identical to the Mn of this sample determined by SEC. Similar calculations for the other PD entries in Table 1 are consistent with this interpretation. In fact, this means that this could lead to a maximum Mn that could be expected using D, as seems to be the case would be about 12 kg mol−1 (100% conversion with no added initiator), and thus more rigorous purification methods would be useful to pursue.
- K. M. Stridsberg, M. Ryner and A. C. Albertsson, Adv. Polym. Sci., 2002, 157, 41–65 CAS.
- H. Tobita and A. E. Hamielec, Polymer, 1990, 31, 1546–1552 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: CMspectra (1H NMR, COSY, HMQC, 13C NMR, DEPT, EI mass spectrum) and assignment of NMR spectra; Dspectra (1H NMR, HMQC, 13C NMR, EI mass spectrum) and assignment of NMR spectra, poly(D-co-CM) data (1H NMR spectra, SEC elugram); epoxidized poly(D-co-CM) data (1H NMR spectrum). See DOI: 10.1039/c0py00283f |
| This journal is © The Royal Society of Chemistry 2011 |