Cyclic polyesters: synthetic approaches and potential applications
Jessica N.
Hoskins
and
Scott M.
Grayson
*
Department of Chemistry, Tulane University, New Orleans, LA 70118, USA. E-mail: sgrayson@tulane.edu
First published on 2nd July 2010
Abstract
The synthetic strategies that have been developed for the preparation of cyclic polyesters are reviewed. While linear and branched polyesters are used extensively for biomedical and materials applications, the synthetic difficulty of preparing cyclic polyesters has limited their exploration. Recently synthetic methodologies have been developed which compliment previous techniques, and promise to increase the availability of cyclic polyesters for future studies. The relative advantages and disadvantages of each approach are discussed, as well as some of the unique physical properties demonstrated by cyclic polymers.
 Scott M. Grayson and Jessica N. Hoskins | Jessica N. Hoskins earned her BS in Biochemistry in 2006 from Grove City College in Grove City, PA where she worked in the lab of Professor Durwood B. Ray. She is currently a Louisiana Board of Regents fellow, pursuing a PhD in Chemistry at Tulane University under the guidance of Prof. Scott M. Grayson. Her research is focused on the synthesis and characterization of novel polymer architectures based on cyclic polyester substrates. |
Scott M. Grayson received a BS from Tulane University (1996) and a PhD in Chemistry from the University of California, Berkeley (2002), studying the role of polymer architecture for drug delivery under Jean M. J. Fréchet. Following post-doctoral studies with C. Grant Willson at the University of Texas, he accepted a faculty position in the Department of Chemistry at Tulane University in New Orleans. After a brief “hurricane sabbatical” he returned to Tulane in 2006 to continue his research, focusing on the synthesis, characterization, and application of complex, yet well-defined polymer architectures with a particular focus on cyclic polymers. |
Introduction
Polyesters were one of the earliest families of polymers explored synthetically, and are also one of the most industrially important classes of polymers. Aliphatic polyesters are a particularly relevant subfamily as many exhibit both biocompatibility and biodegradability, in addition to showing promise as recyclable materials made from renewable bio-feed stock.1 Although condensation polymerizations provide an important route to polyesters, the control of polydispersity and end group functionalities exhibited in ring opening polymerizations has lead to their preferential use for many synthetic explorations. A variety of lactone monomers (Scheme 1) have been investigated extensively for use in an assortment of medical applications, including prosthetics, dental implants, stents, degradable sutures, and drug delivery scaffolds.2–4 Because a polymer's physical properties are inherently tied to its molecular structure, a range of non-linear aliphatic polyester architectures, including star,5–10 graft,11–16 cross-linked,17–19 and hyberbranched20–24polymers have been investigated to address the diverse material and biomedical applications of polyesters. However, despite exhibiting a particularly unique set of physical properties, cyclic polymers are among the least explored class of polyester architectures because of difficulties in both their preparation and their purification.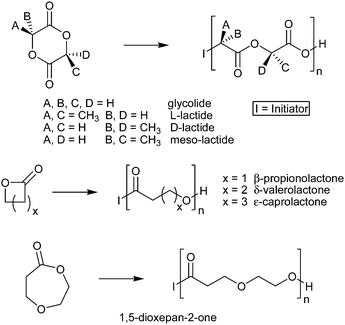 | ||
| Scheme 1 General structures of common aliphatic polyesters. | ||
Cyclic polyesters are of particular interest as cyclic polymers have been shown to possess unique properties when compared to their linear counterparts. The cyclic topology and lack of chain ends impart upon cyclic polymer materials properties that are substantially different than those of their linear analogues, including increased glass transition temperatures (Tg), smaller hydrodynamic volumes, and lower intrinsic viscosities.25 In addition, the cyclic topology imparts unique biophysical properties which may provide advantages for a range of biomedical applications. For example, the biodistribution of cyclic polymers has been demonstrated to be different from that of linear polymers, leading to some potential advantages as novel drug delivery vectors.26,27 In addition, during hydrolytic degradation, the reduction in both mass and hydrodynamic volume is substantially reduced compared to their linear analogs, providing a retarded degradation profile.28 And finally, cyclic polyesters offer some promise as supramolecular hosts. The behavior of cyclodextrins,29–31calixarenes,32,33cucurbiturils,34,35 and crown ethers36–39 provides some foundation for the complexation of guests within smaller cyclic polyesters, and cyclic poly(lactide) hexamers have been shown to exhibit enhanced binding through the synergistic electron donation of the multiple carbonyl functionalities.40 However, to date this has been explored in a very limited fashion for larger polyesters.
While the preparation of high purity cyclic polymers remains a challenge, an increasing number of approaches have been reported recently,41,42 including some which offer compatibility with the chemistry of polyesters. The many routes that have been explored for the preparation of cyclic polyesters can be divided into four categories: the statistical cyclization of linear polyesters during ester condensation polymerizations, the ring expansion from cyclic tin initiators by insertion of lactone monomers, the N-heterocyclic carbene catalyzed cyclopolymerization of lactides, and finally the cyclization of α,ω-functionalized linear polymer precursors under high dilution. Each of these techniques has a unique set of advantages and disadvantages, which will be reviewed below. This review will also attempt to highlight some of the most promising aspects of this area of research, as well as the remaining challenges.
Physical properties
Many of the dilute solution properties and the bulk materials properties of cyclic polymers are significantly different from their linear analogs.42 While these differences can potentially be utilized for materials applications, they are also of significant interest for understanding the fundamental structure/property relationships exhibited by different polymer architectures. In addition, these property differences provide an invaluable characterization tool for differentiating cyclic polymers from linear materials. While most of the initial physical studies on cyclic polymers focused on cyclic polystyrene and other non-degradable backbones, the changes in physical properties which are exhibited by all cyclic polymers appear to be comparable regardless of backbone functionality.Cyclic polymers exhibit a range of unique dilute solution properties, which in essence measure the fundamental physical properties of individual macromolecules, without the influence of intermolecular interactions due to neighboring polymer chains.25 For example, the ratio of the cyclic hydrodynamic radii to the linear hydrodynamic radii (gh = (Rh)c/(Rh)l) is reported to fall between 0.84 and 0.89 for polystyrene, with subtle fluctuations likely caused by differences in the polymer molecular weights or the solvents used.43–46 These values are in agreement with theoretical predictions, as the cyclic architecture reduces the conformational freedom relative to linear polymers with free chain ends. This smaller hydrodynamic radius results in a slower elution time in gel-permeation chromatography (GPC) relative to linear counterparts, and thus is often used to confirm the cyclic structure of a polymer if analogous linear polymers are available for comparison. For the same reason, the radii of gyration also consistently exhibit lower values for cyclic polymers when compared to linear analogs.45,47–49 For polystyrene samples, the ratio of the mean square radii of gyration (〈Rg〉2c/〈Rg〉2l) is found to fall between 0.50 and 0.51. Another well studied dilute solution property is intrinsic viscosity, as this property is easily measured and very sensitive to differences in architecture.25 When comparable pairs of linear and cyclic polystyrene samples were examined, the ratio of the intrinsic viscosities (g′ = [η]c/[η]l) is reported to be between 0.64 and 0.68,50 though again these values seem subject to minor fluctuations with backbone, molecular weight, and solvent.
When examining the bulk properties, the cyclic architecture also has an effect on the glass transition temperature (Tg), the melting temperature (Tm), and the thermostability. Perhaps the most studied comparison of bulk properties between linear and cyclic analogs of polystyrene and polybutadiene is their glass transition temperatures.43,49–52 Because the cyclic polymers lack chain ends, the mobility of any segment resembles the “middle” portion of linear polymers.53 As a result, they exhibit very little change in Tg with respect to molecular weight, in contrast to linear systems.54 The melt transition (Tm) exhibits similar effects with architecture, as does the thermostability, however, the latter is affected strongly by the functionality of end groups that are present. While the majority of the physical properties described above were carried out on cyclic polystyrene, due to its synthetic availability, initial studies of the thermal properties of linear and cyclic polyester analogs agreed with these previously reported trends.25 In the case of thermogravimetric analysis, however, the identity of the end groups appeared to be the most critical factor as mass loss was dominated by competing backbiting and non-backbiting degradation mechanisms.55
A unique aspect of cyclic polyesters, in contrast to the commonly studied cyclic polystyrene, is their facile hydrolytic and enzymatic degradation. A recent study comparing the degradation behavior of cyclic poly(ε-caprolactone) reveals a substantial difference when compared to linear analogs. When examining the mass loss by MALDI-TOF mass spectrometry, poly(ε-caprolactone) cyclics exhibit the expected retardation of mass loss, since the first bond scission along the polymer chain does not reduce the molecular weight. However, when the same degradation is observed using GPC, the cyclic polymers actually exhibit an increased size during the initial phase of their degradation, because their hydrodynamic radius increases during the transformation from a cyclic to a linear architecture that occurs with the first bond scission.28
Perhaps the most interesting properties exhibited by cyclic polymers are their biological properties in vivo. Szoka and co-workers found that the cyclic architecture increases blood circulation times,26 and, in subsequent work, found it also increases the tumor uptake when compared to linear polymers of the same molecular weight.27 Cyclic polyesters were utilized in this initial study largely because their degradability assures their eventual clearance from the body. These combined properties of cyclic polyesters have many important implications for their use as scaffolds in drug delivery and imaging, where longer circulation times can be critical for optimizing targeting. As low PDI's (<1.2) are necessary to achieve meaningful data in these biological applications,56 routes to well-defined cyclics are vital for the viability of these biomedical studies and applications.
Synthesis
1. Statistical cyclization of polyesters during condensation polymerization
Cyclic polyesters were first observed as low molecular weight byproducts of linear polyesters during condensation polymerization; however, the products were inevitably a mixture of linear and cyclic polymers. When carrying out a statistical cyclization, high dilution is critical to favor the formation of cyclic polymers, because such conditions reduce the rate of intermolecular coupling, while the rate of intramolecular cyclization remains unchanged.57–59 While it is possible to increase the purity of the macrocyclic products by utilizing their contrasting physical properties relative to the linear impurities, these techniques are both inefficient and tedious (e.g.fractionation or preparative GPC).An elegant approach to improving the yield of cyclic polyester production during a condensation polymerization involves attaching the propagating polymer to a solid support, such that the primary means of releasing a soluble polymer from the solid phase is through the “backbiting” reaction that only occurs during cyclization. This was first demonstrated by Wood et al., using 11-unbromodecanoic acid monomers complexed to the ammonium cation of Amberlyst A-26 exchange resin with chloride counterion.60 The resin was first washed with aqueous potassium bicarbonate to convert the chloride counterion of the solid phase to bicarbonate, and then the carboxylic acid monomer was introduced, releasing gaseous carbon dioxide, and generating 11-bromoundecanoate anions bound to the solid support. Polymerization of the bound monomer could be activated thermally (55 °C), enabling a carboxylate from one monomer to displace the bromide of an adjacent monomer. Propagation generates increasingly longer linear chains bound by the carboxylate end group, until displacement of the terminal bromide by the carboxylate on the opposite end of the same polymer chain generates an uncharged cyclic polymer, which is then released into the solution phase (Scheme 2). After extracting the cyclic product, the remaining bound linear polymer can be released from the solid phase by reaction with methyl iodide, yielding free linear polyester that could be characterized for comparison with the macrocycles. However, because competing reactions, such as nucleophilic attack by carboxylate or methanol residues, generate linear polymers, the cyclic materials required further purification by preparative GPC, resulting in a series of fractions ranging in number average molecular weight (Mn) from 950 to 2800, each with a low polydispersity index (<1.2). 1H NMR characterization confirmed that the bromide end group was missing from the proposed cyclic fractions and the single distribution observed in the fast atom bombardment (FAB) mass spectra was consistent with a cyclic polymer without end groups.
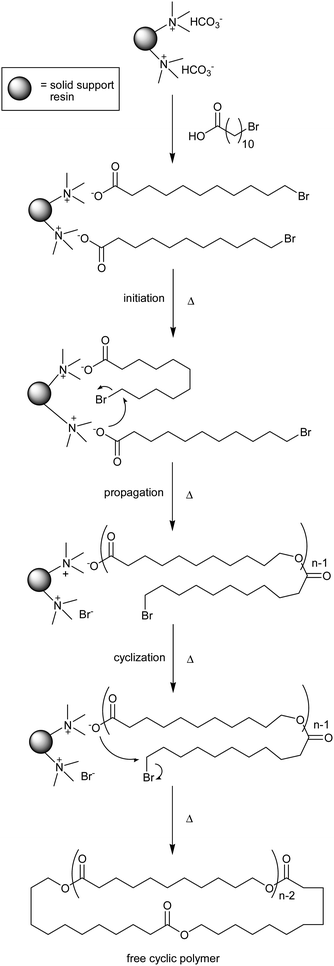 | ||
| Scheme 2 The first synthetic technique designed to produce predominantly cyclic polyesters utilizing a quaternary ammonium functionalized solid support resin.60 | ||
A similar solid phase technique using covalently immobilized monomer was investigated by Ruddick et al. for the production of macrocyclic lactone oligomers.61 The carboxylic acids of the ω-hydroxyacid monomers, such as 11-hydroxyundecanoic acid, were bound to a chloromethylated polystyrene resin viaester linkages. Upon heating (133 °C) in the presence of a catalyst both chain growth and cyclization reactions occur, but again only the cyclization reaction released the polymer from the solid support (Scheme 3). In order to avoid preparative GPC purification, various monomer/initiator combinations were investigated to identify which provided the best purity and highest molecular weight cyclic polymer. The optimized conditions utilized ω-hydroxyacids bearing primary hydroxyl groups, di-n-butyltin oxide as the catalyst and chlorobenzene as solvent to produce the cyclic polyester oligomers as the primary soluble product (>92%). However, the degrees of polymerization generally remained low (<7), as previously observed by Wood et al., due to the inefficient propagation resulting from the limited mobility of the covalently tethered monomer.
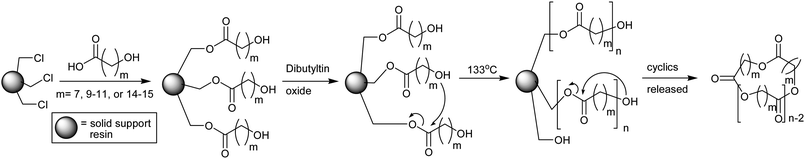 | ||
| Scheme 3 Esterification of chloromethylated polystyrene resin with various ω-hydroxyacids, followed by intermolecular propagation and then intramolecular cyclization to release cyclics into the solution phase.61 | ||
A final example of the formation of cyclic polyesters with the aid of solid support reaction systems is reported by the Chisholm et al.40 In contrast to previous techniques, the initiator was bound to the support so that the monomer could remain in the solution phase. In this reaction the anionic initiator was bound to the solid support resin by reacting amino-functionalized polystyrene resin with n-butyl lithium, Me2Mg, or Et2Zn. The resin was then reacted with a few equivalents of L-lactide (10 per each N) at room temperature, which initiated growth of the PLA oligomers from the solid phase metal amide anion. The activated resin was then reacted with additional L-lactide in benzene at room temperature to extend the PLA oligomers. Upon heating to 70 °C, the rate of the intrachain transesterification (cyclization) is increased, and cyclic oligoesters are released into solution in significant quantities (Scheme 4). The obtained cyclics exhibited a broad polydispersity index (approximately 2.0) and low molecular weights (Mn < 1000). Of the three metal counterions tested, lithium was the most reactive, which is consistent with findings for other homogenous polymerizations in solvents such as benzene.
 | ||
| Scheme 4 Synthesis of metal-functionalized resin used to initiate the polymerization of lactide. Upon heating, intrachain transesterification releases cycles from the solid support. Note that x and y indicate that the intrachain transesterification can occur at any point along the polymer backbone.40 | ||
Interestingly, however, it was found that the release of cycles into solution occurs reversibly, and thus, the rings released in solution remain in dynamic equilibrium with the resin-bound polymer. For example, if NaBPh4 was introduced into the solution phase, cyclic PLA of a certain size could be templated, resulting in an 80% conversion to the macrocycle containing 6 ester linkages.40NaBPh4 is known to bind strongly to these cyclic lactic acid hexamers through a synergistic chelation of the sodium by three of the carbonyl oxygens from every other ester in the macrocycle, and therefore the authors propose that the cyclization was reversible, enabling dynamic selection of the thermodynamically favored complex.
Though, in theory, the polymerization from a solid support appears to be an elegant technique for isolating the desired cyclic polyesters from their inevitable linear byproducts, a number of practical aspects limit its application. The rate of propagation from a solid support is fundamentally slow and non-uniform, resulting in the generation of predominantly oligomeric products with broad polydispersities. Future solid-phase syntheses must overcome this challenge in order to generate truly polymeric cyclics. In addition, very clean propagation and efficient cyclization reactions must be incorporated into the synthetic design to prevent the release of linear polymersvia side reactions, such as interchain transesterification or nucleophilic attack by small molecular weight impurities. Such side reactions have been observed in most of the examples investigated, and essentially negate the primary advantage of the solid-supported polymerization: the ease of purifying the cyclic product. However, this approach has been demonstrated to efficiently prepare high purity, low molecular weight oligomeric macrocycles with the assistance of a templating cation.
2. Ring expansion techniques
An alternative technique developed for the synthesis of cyclic polyesters is broadly classified as a ring expansion polymerization. In ring expansion polymerizations, initiation occurs by the insertion of a cyclic monomer into the relatively labile bond of a cyclic initiator, followed by propagation by analogous insertion of additional monomer. This approach is particularly attractive because, ideally, the synthesis does not involve the generation of linear intermediates, therefore providing an efficient route to pure cyclic polymers. Two distinct variations of this approach have been investigated in detail. In the first, the labile bond in the initiator is a metal–oxygen bond (most commonly Sn–O), and in the second the labile link is an ionic bond between a carboxylate ion and an imidazolium cation, generated by the addition of an N-heterocyclic carbene to the lactone monomer.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 were achieved, marking a substantial increase over previous techniques for the synthesis of cyclic polyesters.
000 were achieved, marking a substantial increase over previous techniques for the synthesis of cyclic polyesters.
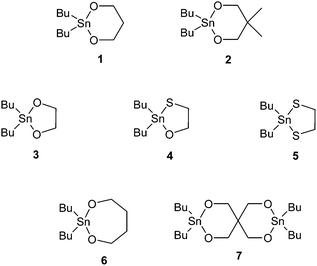 | ||
| Fig. 1 Chemical structures of cyclic tin initiators employed for the cyclopolymerization of various lactones and lactides. | ||
 | ||
| Scheme 5 First example of ring-expansion polymerization of a cyclic polyester viainsertion into a cyclic tin initiator.62 | ||
Because the bond between the initiator and macrocycle is particularly labile, linear analogs can be produced by allowing the polymer to undergo competitive ligand exchange in the presence of ethane-1,2-dithiol62 (Scheme 6). The cleavage was successful due to the increase in stability of Sn–S bonds relative to Sn–O bonds. This exchange aided characterization, as it provides synthetic access to analogous linear polymers with the same Mn and PDI as their cyclic precursors. During size exclusion chromatography, the cyclic polymers exhibited a longer retention time than their linear counterparts because the cyclic confinement leads to a smaller hydrodynamic volume.
 | ||
| Scheme 6 Competitive ligand exchange with ethane-1,2-dithiol and cyclic β-BL to yield analogous linear β-BL samples for comparison with cyclic analogs during physical characterization.62 | ||
In an extensive series of investigations, Kricheldorf and co-workers explore the use of this ring expansion technique to produce other cyclic polyesters. For example, bulk cyclic polymerizations of L-lactide and D,L-lactide were reported at 120 °C using similar cyclic tin initiators 2,2-dibutyl-2-stanna-1,3-dioxane, 2,2-dibutyl-2-stanna-1,3-oxathiolane, and 2,2-dibutyl-2-stanna-1,3-dithiolane (3, 4, and 5 in Fig. 1).63 For both L-lactide and D,L-lactide, polydispersity indices were broad (2.0–3.5). While molecular weights of up to Mn = 110000 were reported, the molecular weights were difficult to control and did not demonstrate “living” character, as the achieved molecular weights did not correlate to those expected for the monomer/initiator ratios used. Also, the rate of propagation was higher than the rate of initiation for both initiators studied. The authors hypothesized that this may be due to the tendency of the initiators used to form dimers, resulting in high melting points and low solubility in organic solvents and the monomer melts.64 The initiator 2,2-dibutyl-2-stanna-1,3-dioxepane (DSDOP) (6 in Fig. 1) is a monomeric liquid miscible with organic solvents and lactones, and Kricheldorf and Eggerstedt report its use for the polymerizaton of cyclic poly(ε-caprolactone) and cyclic poly(β-butyrolactone) in bulk at 80 °C with lower polydispersities (1.5) and molecular weights from Mn = 5800 to 126000. While the propagation step was still faster than the initiation, the observed Mn values did correlate closely to the monomer/initiator ratio, as would be expected for a living polymerization.
Stridsberg and Albertsson used the cyclic tin initiator 2,2-dibutyl-2-stanna-1,3-dioxane (3 in Fig. 1) for the polymerization of cyclic 1,5-dioxepan-2-one.65 While in previous work, cyclic ring expansion polymerizations were conducted in bulk at high temperatures (>70 °C), Stridsberg and Albertsson found that using a cosolvent (methylene chloride or chloroform) and lower temperature (30 °C) slowed the polymerization, yielding similar molecular weights (up to Mn = 29000) but with significantly reduced polydispersity indices (as low as 1.34). However, the authors were unable to obtain bulk polymerization results with their system for comparison because of the poor solubility of the metal initiator in the monomer melt.
In addition, this technique is amenable to making more complex architectures, and more diverse chemistries, with the use of appropriate initiators. For example, the use of a spiro-bicyclic distannoxane initiator (7 in Fig. 1) for the ring expansion of ε-caprolactone yields figure-eight shaped polymers (Fig. 2).66Sulfur containing polymers could also be prepared by substituting the lactone monomers with γ-thiobutyrolactone and ε-thiocaprolactone.67 This synthetic technique also provides unique routes to telechelic, star-shaped, diblock, and triblock polyesters.68
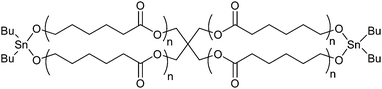 | ||
| Fig. 2 Figure-eight shaped poly(ε-caprolactone) synthesized from a spiro-bicyclic distannoxane initiator.66 | ||
However, the retention of the tin initiator as part of the macrocycle is problematic for several reasons. The lability of the bond between the initiator and macrocycle makes it susceptible to alcoholysis or hydrolysis under mild conditions, and also makes it susceptible to fragmentation during mass spectral analysis, limiting the methods available for characterizing these materials. More importantly, the toxicity associated with the tin initiator that is retained within the cyclic polymer limits the potential in vivo applications of otherwise biocompatible and biodegradable polyesters.
To address these problems, Kricheldorf and co-workers developed a technique in which the labile linkages from the tin initiator could be replaced with a hardier 1,3-dithian-2-one linkage.67 The poly(ε-caprolactone) macrocycle undergoes a ring insertion/ring elimination process, such that both of the alkoxide ligands of the tin initiator are exchanged with thiolates from dithianone without ring opening of the macrocycle (Scheme 7).
 | ||
| Scheme 7 Competitive ligand exchange for the synthesis of stabilized, tin-free poly(ε-caprolactone) macrocycles.67 | ||
Alternatively, Liet al. report a novel approach to ring stabilization by polymerizing a few repeat units of a photo-crosslinkable lactone block of α-(1-acryloxyethyl)-ε-caprolactone after the initial cyclic PCL block initiated by DSDOP (6 in Fig. 1).69 The acrylate side chains can be cross-linked by UV under dilute conditions to form a more stabile cyclic link. Afterwards, the tin alkoxide initiator can be intentionally hydrolyzed from the macrocycle (Scheme 8). Liet al. also report that the polymerization of ε-caprolactone can be resumed after UV treatment but prior to hydrolysis of the tin initiator, providing a unique route to double-tailed tadpole-shaped copolyesters.
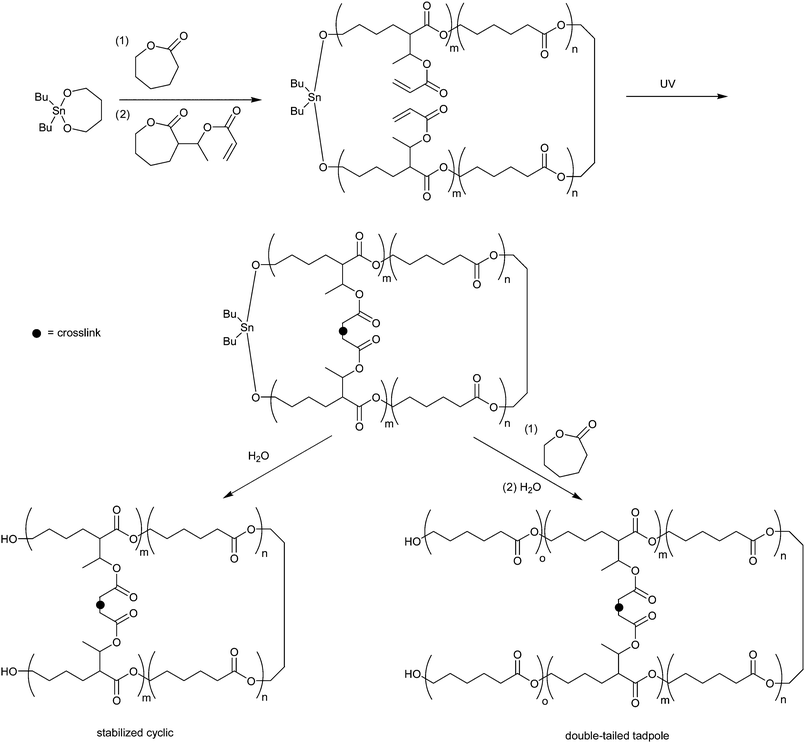 | ||
| Scheme 8 Synthetic strategy for the synthesis of stabilized poly(ε-caprolactone) diblock macrocycles and tadpole shaped triblock polymers free from tin initiator.69 | ||
As the polyester backbone is susceptible to cleavage under basic, acidic, and nucleophilic conditions, the incorporation of side-chain functionalities is particularly difficult. However, a couple of examples have been demonstrated from this ring expansion approach. For example, Liet al. report the incorporation of α-chloro-ε-caprolactone and ε-caprolactone copolymers into the “tails” of the double-tailed tadpole copolyesters previously reported.70 The pendant chloride groups were then converted to azides, and alkynyl functionalized PEO (Mn = 1000) was grafted to the tails by copper-mediated Huisgen cycloaddition (“click” reaction) to yield amphiphilic tadpole-shaped poly(ε-caprolactone) grafted with PEO. Additionally, Liet al. report the synthesis of cyclic copolymers of ε-caprolactone and 5-triethylsilyloxy-oxepan-2-one.71 A few units of α-(1-acryloxyethyl)-ε-caprolactone were polymerized at the end of the macrocycle and cross-linked to retain its integrity. The triethylsilanolate groups were deprotected (without significant degradation of the polymer chain) by dropwise addition of 40% hydrofluoric acid to the polymer dissolved in acetonitrile to reveal pendant alcohol groups for further functionalization of the cyclic. Poly(ethylene oxide) (PEO) monocarboxylic acid (Mn = 1050) was grafted onto the cyclics by N,N′-dicyclohexylcarbodiimide (DCC) esterification to produce amphiphilic PEO grafted macrocycles of modest molecular weights (Mn = 24000) and relatively low PDI (1.4). The grafting efficiency was estimated to be 50–60%, and the cyclics were composed of an average of 7–8 PEO chains per 165 repeat unit macrocycle. This technique also allows the synthesis of unique architectures, such as twin double-tailed tadpoles.72
The dioxastannane cyclic ring expansion approach exhibits substantial advantages over the previously reported cyclization approaches. As long as care is taken to remove any linear monomers or initiators, the purity of the cyclic product is exceptional, circumventing the need for additional purification. In addition, because the synthetic pathway does not involve linear intermediates, complications with physical cross-links or knots due to entanglement are avoided that might impact the macroscopic properties of macrocycles.44 The efficiency of the solution-phase polymerization also enables access to high molecular weight polymers (>100000). However, the polydispersities obtained tend to be moderate to high (1.2–2.5) and the need for stabilization of the labile tin linkage requires additional synthetic steps.
000 with narrow polydispersities (<1.35). In the initiating step, the NHC (in this case, 1,3-dimesitylimidazol-2-ylidene (IMes)) attacks the carbonyl of the lactide to produce a zwitterionic alkoxide/imidazolium ion pair. The alkoxide can then carry out a nucleophilic attack of additional monomer to expand the poly(lactide) tether between the ion pair. Finally, the proximity of the alkoxide ion to the opposite end of the polymer chain encourages a backbiting reaction to yield cyclic poly(lactide) and a regenerated NHC catalyst (Scheme 9). Because the rates of propagation and cyclization are inherent to the catalyst and monomer in the system, the molecular weight range of the polymerization is largely defined by the selected catalyst and monomer.
 | ||
| Scheme 9 Zwitterionic N-heterocyclic carbene catalyzed ring expansion cyclopolymerization of lactide.76 | ||
In addition to possessing narrow polydispersities, these cyclics were produced extremely rapidly (<120 s) at relatively high monomer concentrations (0.6–1.0 M in THF). Furthermore, unlike most of the dioxastannane catalyzed ring expansion polymerizations, NHC-mediated polymerizations display a remarkable degree of control and exhibit some features of living polymerizations, such as linear plots of molecular weight (Mn) against monomer conversion and polydispersity indices less than 1.3.76 Similarly, however, the initiation rate was found to be slower than the rate of propagation. The cyclic nature of these poly(lactide)s was confirmed by the absence of end groups in the MALDI-TOF mass spectrum. While exactly analogous linear polymers could not be easily prepared from the cyclic polymer, linear PLA samples of similar molecular weight (as determined by light scattering) were separately polymerized from benzyl alcohol initiators. When compared to their cyclic counterparts of similar molecular weight, the linear samples exhibited a larger apparent molecular weight (by GPC), a higher intrinsic viscosity, and a lower thermal stability, providing further evidence for the cyclic nature of the product. The cyclopolymerization of β-butyrolactone and β-propiolactone catalyzed by the saturated NHC 1,3-dimesitylimidazolin-2-ylidene (SIMes) was also reported for molecular weights of up to Mn = 5000 and with similar PDI < 1.37.77
In subsequent publications, Waymouth and co-workers performed mechanistic and kinetic studies on the cyclization reactions catalyzed by the NHC IMes.78 These studies reveal that the rate of initiation is second order in monomer concentration, and slower than the propagation. However, the rate of propagation is significantly faster than the rate of cyclization. Together, these traits account for the observed narrow molecular weight distribution, as well as the limits that the cyclization step places on the molecular weights that can be achieved with this particular catalyst. However, initial reports of the synthesis of poly(ε-caprolactone) using a less hindered catalyst, 1,3,4,5-tetramethylimidazol-2-ylidene, yielded cyclic polymer with molecular weights above 100000.79
The NHC-catalyzed ring expansion polymerization provides a rapid route to the most well defined cyclic polymers in the intermediate molecular weight range (10000–50000), however, additional catalyst optimization will be required to access higher molecular weight cyclic polymers. Because the attainable molecular weight is dependent upon the inherent ratio of the rates of propagation and cyclization for a specific catalyst/monomer system, investigations of alternative catalytic NHCs offer promise to access higher molecular weights. However, for investigations related to the physical properties of the cyclic polyesters, the inability to make an exactly analogous linear polyester makes it more difficult to isolate the effects of the cyclic architecture. In addition, side chain functionalized polymers have yet to be reported with this approach, and the carbene catalyst is expected to exhibit only modest compatibility with many desirable side chain functionalities.
3. α,ω-Ring closure
A final approach for the production of cyclic polyesters involves the cyclization of α,ω-functionalized linear precursors under dilute conditions. For this approach it is critical that the cyclization occurs under high dilution, so that the linear precursors favor intramolecular cyclization over intermolecular oligomerization. Therefore the coupling reaction must be extremely efficient to generate high purity macrocycles. In an adaptation of the copper catalyzed azide–alkyne click technique first reported for the synthesis of cyclic polystyrene,80 Hoskins and Grayson first prepared linear poly(ε-caprolactone) using 3-azidopropanol as the initiating species and tin(II) 2-ethylhexanoate as the catalyst.28,81 The terminal alcohol on the linear PCL chain was then esterified with pentynoic anhydride to provide complimentary azide and alkyne functionalities on opposite ends of the linear precursor. In the presence of a Cu(I) catalyst, the rate of the 1,3-dipolar cycloaddition reaction between the alkyne and azide groups is sufficiently rapid82–85 that “pseudo-high dilution” can be achieved by adding the precursor dropwise into a solution of the catalyst. The rate of addition can be tuned empirically such that the rate of dropwise addition is slower than the rate of the cyclization. This maintains an extremely low precursor concentration throughout the coupling reaction and yields extremely pure cyclic with negligible linear or oligomeric byproducts (Scheme 10). The use of a primary alcohol initiator and tin(II) 2-ethylhexanoate catalyst for the ring opening polymerization allows the production of linear precursors with very narrow polydispersities (<1.2) in the molecular weight range between 4000 and 15000. Because the cyclization reaction is nearly quantitative, the only post-cyclization purification required is an aqueous wash to remove the copper catalyst from the cyclic product.
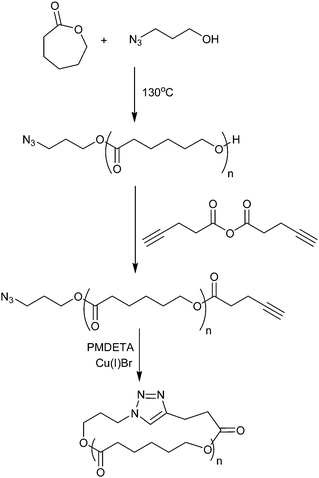 | ||
| Scheme 10 Synthesis of cyclic poly(ε-caprolactone) via a combination of ring opening polymerization and “click” chemistry.28,81 | ||
The same “click” ring closing approach was utilized by Misaka et al. for the polymerization of δ-valerolactone initiated by 6-azide-1-hexanol and catalyzed by 1,8-diazabicyclo-[5.4.0]undec-7-ene (DBU) and 1-[3,5-bis(trifluoromethyl)phenyl]-3-cyclohexylthiourea (BCT), yielding cyclic material with comparable molecular weights (Mn = 2600–10000) and polydispersity indices (<1.2).86 Xie et al. examined a variety of catalytic coupling reactions for the ring closure of poly(ε-caprolactone), including ring closing metathesis, ring closing enyne metathesis, and the “click” reaction to determine which was the most efficient for generating pure cyclic polymers.87 Of the three coupling reactions, the “click” cyclization yielded an exceptionally pure cyclic product, whereas both of the ring closing metathesis cyclization reactions produced significant amounts of intermolecular dimers and trimers when carried out under similar dilution. “Click” ring closure has also been utilized to produce figure-eight shaped copolymers of poly(ε-caprolactone) and polystyrene.88
The cyclization of α,ω-functionalized linear precursors is particularly versatile for incorporating diversity into the backbone of the polymer, because the backbone can be generated from a wide variety of monomers and catalysts while the click coupling reaction is orthogonal to a wide range of side chain functionalities. Since the linear precursors are prepared first, polymerization catalysts already optimized for linear polymerizations can be used to produce well defined precursor material. The most substantial drawbacks to this technique are a consequence of the post-polymerization cyclization requirements. Because the cyclization must occur under high dilution to give pure cyclic, the generation of polymer is typically carried out on small scales. Furthermore, the difficulty of both quantitative end-group functionalization and efficient α,ω-cyclic coupling for significantly larger polymers limits the size of polymer macrocycles that can be generated using this technique.
Conclusion and outlook
Within the last few years, a number of complementary synthetic methodologies have been developed for preparing cyclic polyesters. While cyclic polymers have been studied because of their unique physical properties and their “endless” topological features,89 perhaps the most intriguing potential applications, in the form of encapsulation and drug delivery, remain relatively unexplored. As low PDI's (<1.2) are necessary for polymeric materials used as a therapeutic in vivo, it is reasonable to hypothesize that NHC catalyzed ring expansion polymerizations and α,ω-functionalized ring closure techniques are most amenable for future studies, with the former being most appropriate for moderate to high molecular weight polyesters. Furthermore, the ability to couple functionality onto the side chains of polyesters has been explored in detail for linear systems and could be easily applied to cyclic polyesters to enable the tethering of drugs, solubilizing groups, or targeting moieties. In this regard, the functional group compatibility of the click cyclization approach is particularly appealing. While linear and dendritic polyesters are the subject of increasingly advanced polymer–drug conjugate research, it is expected that the cyclic polyesters will exhibit additional unique and useful properties that will undoubtedly soon be discovered.Acknowledgements
The authors thank the Louisiana Board of Regents for a graduate fellowship, as well as the American Chemical Society Petroleum Research Fund (ACS-PRF 47108-G7), and the National Science Foundation (NSF CAREER-ARRA 0844662) for financial support.Notes and references
- C. K. Williams and M. A. Hillmyer, Polym. Rev., 2008, 48, 1–10 Search PubMed.
- M. Vert, S. M. Li, G. Spenlehauer and P. Guerin, J. Mater. Sci.: Mater. Med., 1992, 3, 432–446 CrossRef CAS.
- A.-C. Albertsson and I. K. Varma, Biomacromolecules, 2003, 4, 1466–1486 CrossRef CAS.
- P. A. Gunatillake and R. Adhikari, Eur. Cells Mater., 2003, 5, 1–16 CAS.
-
M. Spinu, US Pat., 5225521, 1993.
- S. H. Kim, Y.-K. Han, K.-D. Ahn, Y. H. Kim and T. Chang, Makromol. Chem., 1993, 194, 3229–3236 CrossRef CAS.
- D. Tian, Ph. Dubois, R. Jérôme and Ph. Teyssié, Macromolecules, 1994, 27, 4134–4144 CrossRef CAS.
- M. Trollsås, J. L. Hedrick, D. Mecerreyes, Ph. Dubois, R. Jérôme, H. Ihre and A. Hult, Macromolecules, 1997, 30, 8505–8511 CrossRef.
- J. L. Hedrick, M. Trollsås, C. J. Hawker, B. A. Atthoff, H. Claesson, A. Heise and R. D. Miller, Macromolecules, 1998, 31, 8691–8705 CrossRef CAS.
- S. Hecht, H. Ihre and J. M. J. Fréchet, J. Am. Chem. Soc., 1999, 121, 9239–9240 CrossRef CAS.
- T. C. Chung and D. Rhubright, Macromolecules, 1994, 27, 1313–1319 CrossRef CAS.
- E.-J. Choi, C.-H. Kim and J.-K. Park, Macromolecules, 1999, 32, 7402–7408 CrossRef CAS.
- R. Gref, J. Rodrigues and P. Couvreur, Macromolecules, 2002, 35, 9861–9867 CrossRef CAS.
- B. Parrish and T. Emrick, Macromolecules, 2004, 37, 5863–5865 CrossRef CAS.
- B. Parrish, R. B. Breitenkamp and T. Emrick, J. Am. Chem. Soc., 2005, 127, 7404–7410 CrossRef CAS.
- B. M. Cooper and T. Emrick, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 7054–7065 CrossRef CAS.
- T. Aoyagi, F. Miyata and Y. Nagase, J. Controlled Release, 1994, 32, 87–96 CrossRef CAS.
- J. T. Wiltshire and G. G. Qiao, Macromolecules, 2006, 39, 4282–4285 CrossRef CAS.
- T. Muroya, K. Yamamoto and T. Aoyagi, Polym. Degrad. Stab., 2009, 94, 285–290 CrossRef CAS.
- C. J. Hawker, R. Lee and J. M. J. Fréchet, J. Am. Chem. Soc., 1991, 113, 4583–4588 CrossRef CAS.
- M. Johansson, E. Malmström and A. Hult, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 619–624 CrossRef CAS.
- M. Trollsås, B. Atthoff, H. Claesson and J. L. Hedrick, Macromolecules, 1998, 31, 3439–3445 CrossRef.
- X. Yu, J. Feng and R. Zhuo, Macromolecules, 2005, 38, 6244–6247 CrossRef CAS.
- S. Chen, X.-Z. Zhang, S.-X. Cheng, R.-X. Zhuo and Z.-W. Gu, Biomacromolecules, 2008, 9, 2578–2585 CrossRef CAS.
- J. A. Semlyen, Cyclic Polymers, Kluwer Academic, Dordrecht, The Netherlands, 2nd edn, 2000 Search PubMed.
- N. Nasongkla, B. Chen, N. Macaraeg, M. E. Fox, J. M. J. Fréchet and F. C. Szoka, J. Am. Chem. Soc., 2009, 131, 3842–3843 CrossRef CAS.
- B. Chen, K. Jerger, J. M. J. Fréchet and F. C. Szoka, J. Controlled Release, 2009, 140, 203–209 CrossRef CAS.
- J. N. Hoskins and S. M. Grayson, Macromolecules, 2009, 42, 6406–6413 CrossRef CAS.
- M. E. Brewster and T. Loftsson, Adv. Drug Delivery Rev., 2007, 59, 645–666 CrossRef CAS.
- L. Jun and L. X. Jun, Adv. Drug Delivery Rev., 2008, 60, 1000–1017 CrossRef CAS.
- A. Harada, Y. Takashima and H. Yamaguchi, Chem. Soc. Rev., 2009, 38, 875–882 RSC.
- V. Boehmer, Angew. Chem., Int. Ed. Engl., 1995, 34, 713–745 CrossRef.
- J. Rebek, Chem. Commun., 2000, 637–643 RSC.
- J. Lagona, P. Mukhopadhyay, S. Chakrabarti and L. Isaacs, Angew. Chem., Int. Ed., 2005, 44, 4844–4870 CrossRef CAS.
- K. Kim, N. Selvapalam, Y. H. Ko, K. M. Park, D. Kim and J. Kim, Chem. Soc. Rev., 2007, 36, 267–279 RSC.
- C. J. Pedersen, J. Am. Chem. Soc., 1967, 89, 7017–7036 CrossRef CAS.
- C. J. Pedersen, Angew. Chem., Int. Ed. Engl., 1988, 27, 1021–1027 CrossRef.
- J.-P. Collin, C. Bietrich-Buchecker, P. Gavina, M. C. Jimenez-Molero and J.-P. Sauvage, Acc. Chem. Res., 2001, 34, 477–487 CrossRef CAS.
- M. Kralj, L. Tušek-Božić and L. Frkanec, ChemMedChem, 2008, 3, 1478–1492 CrossRef CAS.
- M. H. Chisholm, J. C. Callucci and H. Yin, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15315–15320 CrossRef CAS.
- B. A. Laurent and S. M. Grayson, Chem. Soc. Rev., 2009, 38, 2202–2213 RSC.
- H. R. Kricheldorf, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 251–284 CrossRef CAS.
- J. Roovers, J. Polym. Sci., Polym. Phys. Ed., 1985, 23, 1117–1126 CrossRef CAS.
- G. Hadziioannou, P. M. Cotts, G. ten Brinke, C. C. Han, P. Lutz, C. Strazielle, P. Rempp and A. J. Kovacs, Macromolecules, 1987, 20, 493–497 CrossRef.
- M. Duval, P. Lutz and C. Strazielle, Makromol. Chem. Rapid Commun., 1985, 6, 71–76 CrossRef CAS.
- J. Huang, C. Li and B. He, Makromol. Chem., 1986, 187, 149–155 CrossRef CAS.
- M. Ragnetti, D. Geiser, H. Höcker and R. C. Oberthur, Makromol. Chem., 1985, 186, 1701–1709 CrossRef CAS.
- K. Ishizu and H. Kanno, Polymer, 1996, 73, 1487–1492 CrossRef.
- M. Antonietti and K. J. Folsch, Makromol. Chem., Rapid Commun., 1988, 9, 423–430 CrossRef CAS.
- G. B. McKenna, G. Hadziioannou, P. Lutz, G. Hild, C. Straziell, C. Straupe, P. Rempp and A. J. Kovacs, Macromolecules, 1987, 20, 498–512 CrossRef CAS.
- J. Roovers, Macromolecules, 1988, 21, 1517–1521 CrossRef CAS.
- G. B. McKenna, B. J. Hostetter, N. Hadjichristidis, L. J. Fetters and D. J. Plazek, Macromolecules, 1989, 22, 1834–1852 CrossRef CAS.
- P. G. Santangelo, C. M. Roland, T. Chang, D. Cho and J. Roovers, Macromolecules, 2001, 34, 9002–9005 CrossRef CAS.
- J. Roovers, Macromolecules, 1985, 18, 1359–1361 CrossRef CAS.
- O. Persenaire, M. Alexandre, P. Degée and P. Dubois, Biomacromolecules, 2001, 2, 288–294 CrossRef CAS.
- E. R. Gillies, E. Dy, J. M. J. Fréchet and F. C. Szoka, Mol. Pharmacol., 2005, 2, 129–138 CrossRef CAS.
- H. Jacobsen, C. O. Beckmann and W. H. Stockmayer, J. Chem. Phys., 1950, 18, 1607–1612 CAS.
- D. R. Cooper and J. A. Semlyen, Polymer, 1973, 14, 185–192 CrossRef CAS.
- F. R. Jones, L. E. Scales and J. A. Semlyen, Polymer, 1974, 15, 738–742 CrossRef CAS.
- B. R. Wood, P. Hodge and J. A. Semlyen, Polymer, 1993, 34, 3052–3058 CAS.
- C. L. Ruddick, P. Hodge, A. Cook and A. J. McRiner, J. Chem. Soc., Perkin Trans. 1, 2002, 629–637 RSC.
- H. R. Kricheldorf and S.-R. Lee, Macromolecules, 1995, 28, 6718–6725 CrossRef CAS.
- H. R. Kricheldorf and S.-R. Lee, Macromolecules, 1996, 29, 1375–1381 CrossRef CAS.
- H. R. Kricheldorf and S. Eggerstedt, Macromol. Chem. Phys., 1998, 199, 283–290 CAS.
- K. Stridsberg and A.-C. Albertsson, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 3407–3417 CrossRef CAS.
- H. R. Kricheldorf and S.-R. Lee, Macromolecules, 1996, 29, 8689–8695 CrossRef CAS.
- H. R. Kricheldorf, S.-R. Lee and N. Schittenhelm, Macromol. Chem. Phys., 1998, 199, 273–282 CAS.
- Reviewed in H. R. Kricheldorf, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 4723–4742 Search PubMed.
- H. Li, A. Debuigne, R. Jérome and P. Lecomte, Angew. Chem., Int. Ed., 2006, 45, 2264–2267 CrossRef CAS.
- H. Li, R. Riva, R. Jérôme and P. Lecomte, Macromolecules, 2007, 40, 824–831 CrossRef CAS.
- H. Li, R. Jérôme and P. Lecomte, Macromolecules, 2008, 41, 650–654 CrossRef CAS.
- H. Li, R. Riva, H. R. Kricheldorf, R. Jérôme and P. Lecomte, Chem.–Eur. J., 2008, 14, 358–368 CrossRef CAS.
- M. Swarc, Makromol. Chem., 1960, 35, 132–158 CrossRef.
- G. W. Nyce, T. Glauser, E. F. Connor, A. Möck, R. M. Waymouth and J. L. Hedrick, J. Am. Chem. Soc., 2003, 125, 3046–3056 CrossRef CAS.
- S. Csihony, D. A. Culkin, A. C. Sentman, A. P. Dove, R. M. Waymouth and J. L. Hedrick, J. Am. Chem. Soc., 2005, 127, 9079–9084 CrossRef CAS.
- D. A. Culkin, W. Jeong, S. Csihony, E. D. Gomez, N. P. Balsara, J. L. Hedrick and R. M. Waymouth, Angew. Chem., Int. Ed., 2007, 46, 2627–2630 CrossRef CAS.
- W. Jeong, J. L. Hedrick and R. M. Waymouth, J. Am. Chem. Soc., 2007, 129, 8414–8415 CrossRef CAS.
- W. Jeong, E. J. Shin, D. A. Culkin, J. L. Hedrick and R. M. Waymouth, J. Am. Chem. Soc., 2009, 131, 4884–4891 CrossRef CAS.
- W. Jeong, J. L. Hedrick and R. M. Waymouth, PMSE Prepr., 2009, 101, 1154–1155 Search PubMed.
- B. A. Laurent and S. M. Grayson, J. Am. Chem. Soc., 2006, 128, 4238–4239 CrossRef CAS.
- J. N. Hoskins and S. M. Grayson, PMSE Prepr., 2008, 98, 480–481 Search PubMed.
- N. V. Tsarevsky, B. S. Sumerlin and K. Matyjaszewski, Macromolecules, 2005, 38, 3558–3561 CrossRef CAS.
- H. Gao and K. Matyjaszewski, J. Am. Chem. Soc., 2007, 129, 6633–6639 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef CAS.
- H. Misaka, R. Kakuchi, C. Zhang, R. Sakai, T. Satoh and T. Kakuchi, Macromolecules, 2009, 42, 5091–5096 CrossRef CAS.
- M. Xie, J. Shi, L. Ding, J. Li, H. Han and Y. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3022–3033 CrossRef CAS.
- G.-Y. Shi, L.-P. Yang and C.-Y. Pan, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6496–6508 CrossRef CAS.
- T. McLeish, Science, 2002, 297, 2005–2006 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |