Design of complex polymeric architectures and nanostructured materials/hybrids by living radical polymerization of hydroxylated monomers†
Hamilton
Kakwere
and
Sébastien
Perrier
*
Key Centre for Polymers and Colloids, School of Chemistry, The University of Sydney, Building F11, NSW 2006, Australia. E-mail: sebastien.perrier@sydney.edu.au; Fax: +61 2 9351 3329; Tel: +61 2 9351 3366
First published on 6th August 2010
Abstract
We review the synthesis and application of polymers produced from hydroxyethyl acrylate (HEA) and hydroxyethyl methacrylate (HEMA). Since the first reports in the 1960's on the use of HEMA to produce hydrogels, HEA and HEMA have been extensively used to generate a variety of polymeric architectures and nanostructured materials. In this review, we cover the various conditions of polymerization (monomer purification, solvent, polymer characterization and temperatures), and we specifically look at the use of living radical polymerization to generate well-defined polymers. We then focus on the materials obtained from HEMA and HEA, by considering micelles and nanoparticles, polymeric architectures, hybrid structures and gels.
 Hamilton Kakwere | Hamilton Kakwere obtained his BSc(Hons.) degree in Applied Chemistry in 2002 from the National University of Science and Technology in Zimbabwe. In 2004, he moved to the University of Leeds in UK where he studied for an MSc degree in Polymer and Surface Coatings Science and Technology graduating with distinction in 2005. He recently completed his PhD studies in polymer chemistry at the University of Sydney under the supervision of A/Prof. Sébastien Perrier. His PhD studies focused on the synthesis and self assembly of complex polymeric architectures and macromolecular chimeras. He is currently a postdoctoral fellow at Wageningen University. |
 Sébastien Perrier | A/Professor Sébastien Perrier graduated with a PhD in 2002 from the University of Warwick, after one year as a postdoctoral fellow at the University of New South Wales, he was appointed as a lecturer at the University of Leeds. He moved to the University of Sydney in 2007, and was appointed as director of the Key Centre for Polymers & Colloids. A/Prof Perrier has published over 70 peer reviewed research papers and book chapters. Awards include the Macro Group UK Young Researcher Award (2006), the Young Tall Poppy Science Award (2009), the Rennie Memorial Medal (2009) and the David Sangster Polymer Science and Technology Award (2009). His research interests lie at the interface of polymer synthesis, materials and biology. |
Introduction
Recent developments in organic and polymer synthesis have catalysed a rapid expansion of the production of synthetic polymers. The need for functional materials has triggered the synthesis of an increasing number of functional monomers, among which monomers carrying reactive functional groups have attracted a lot of interest. Indeed, reactive functional groups can be reacted with specific molecules as a means of improving the material's applicability.1–3 For instance, enzymes or drugs can be attached to polymersvia their reactive moieties to impart bioactivity or drug delivery capacity.2Herein, focus is placed on two similar and widely used hydroxyl functionalised polymers, poly(hydroxy ethyl acrylate) (P(HEA)) and its methacrylate analogue, poly(hydroxy ethyl methacrylate) (P(HEMA)) (Fig. 1). The hydroxyl groups are attractive as a functionality in polymers because: (i) they can undergo a wide variety of reactions, (ii) they offer hydrophilic properties and (iii) they can also act as initiation sites for ring-opening polymerization (ROP) of cyclic esters thus, allowing easy access to complex biodegradable polymers. It should be noted here that for these polymers, hydrophilicity does not imply water solubility, since P(HEA) polymers are generally water soluble whilst P(HEMA) is only water soluble at low molecular weight (degree of polymerization (DP) < 20).4
 | ||
| Fig. 1 Chemical structures of P(HEMA) and P(HEA). | ||
Since Wichterle and Lim5 reported P(HEMA) can be used to make hydrogels, this polymer has attracted a lot of interest and has found a wide range of uses in the biomedical field, owing to its biocompatibility and lack of toxicity. P(HEMA) has been widely used to make contact lenses, intraocular lenses, dental fillings, surgical implants, tissue engineering scaffolds, biosensors, prosthetic vascular implants, catheters, hemodialysis membranes, wound dressings and for the immobilisation of drugs, cells and enzymes.6,7P(HEA) is also of interest as it has similar biocompatibility, cytotoxicity, low thrombogenicity and cell compatibility to the widely studied P(HEMA). Consequently, hydrogels of P(HEA) and its copolymers have also been used for making a wide range of biomaterials including hydrophilic sponges, contact lenses, intraocular lenses and enzyme immobilisation.8–10P(HEA) is also widely used in personal care products and surface coatings.
Given that the direct polymerization of HEA/HEMA by ionic polymerizations is not possible due to interaction of the hydroxyl groups with the catalyst and the initiator,2 most of the polymers of P(HEA) and P(HEMA) reported before the late 90s were synthesised via conventional free radical polymerization (in solution) which produces ill-defined polymers. Thus, the synthesis of well-defined P(HEMA)/P(HEA) and their block copolymers before the emergence of living radical polymerization (LRP) techniques has been a major challenge. However, the development of LRP techniques over the years have allowed for the synthesis of well-defined polymers from a wide variety of functional monomers including HEMA/HEA. Thus the construction of block copolymers, star (co)polymers, comb (co) polymers and other complex polymeric architectures of P(HEA)/P(HEMA) is now possible via these techniques. This literature review will draw attention to the use of LRP techniques in the (co)polymerization of HEMA/HEA and the application of their corresponding hydroxylated functional (co)polymers in the synthesis of complex polymeric architectures as well as nanostructured polymeric materials/hybrids. LRP techniques have significantly widened the scope of application of P(HEMA) and P(HEA). For instance, the design of nanostructured polymeric materials such as micelles/nanoparticles has had a major impact in nanotechnology, drug delivery and other many areas of science.11 At the same time, complex polymeric architectures such as dendrimers, branched, star and comb (co)polymers have received a lot of interest over the years due to the differences in properties and behavior of these polymeric architectures compared to simple linear polymers.12,13 Another class of materials of great potential is that of hybrid materials, which comprise of a synthetic polymer joined to another type of material such as carbon nanotube, metal, silica, peptide, protein or DNA.14–16 Such hybrid materials are attractive in that they merge properties of an inorganic or organic naturally occurring material with those of synthetic polymers to form novel (nano)materials with a wide range of potential applications.
Polymer synthesis and characterization considerations
Monomer purification
Commercial HEA/HEMA contains some impurities in the form of di(meth)acrylates, (meth)acrylic acid and ethylene glycol.17 The presence of such impurities in the monomer can have negative impact on polymerizations if they are not removed. For instance, (meth)acrylic acid can slow down/inhibit ATRP polymerizations of HEA/HEMA through coordination of the catalyst with the acid.17 Di(meth)acrylates can lead to formation of cross-linked or branched products which can have an impact on the observed PDIs and product solubility.18 Thus it is prudent to purify monomers prior to polymerization. The method generally used for purifying HEA/HEMA involves washing the monomer with hexane to remove di(meth)acrylates, ether extraction from an aqueous phase to remove acrylic acid then finally vacuum distillation.19 However, Zhang et al.20 have reported the RAFT polymerization of HEA which has not been subjected to this rigorous purification proceeds with good control being observed. Based on the quality of their results, they concluded that purification of HEA by passing through basic alumina is sufficient for RAFT polymerization.Solvents
Polymerization of HEA/HEMA generally requires solvents of high polarity such as DMSO, DMAc, DMF, t-butanol, methanol, water, ethanol or a mixture of polar solvents. Most low polarity solvents used in polymerization of non-polar monomers can dissolve the monomer however the polymer crashes out of the solution when chain lengths increase. The need for polar solvents in HEA polymerization is also applicable when making diblock copolymers where HEA is the second of the blocks.Reaction temperatures
The polymerization temperatures employed for HEMA/HEA with the various polymerization techniques are given in Table 1. The temperatures supplied are from early reports as most works thereafter generally follow similar conditions. Variations from these values tend to be influenced by the extent of dilution and in some cases the type of initiator used.Characterization
SEC is the most common technique used to characterise molecular weight of polymers. Most SEC analyses of P(HEA) homopolymers and block copolymers require the use of polar solvents such as DMF or DMAc with a small amount of LiBr salt to destabilise hydrogen bonding interactions between the polymer chains.21 When used without LiBr, peak distortion is observed.22 Since these are high viscosity solvents, the analysis tends to be done at high temperatures to allow reasonable flow rates to be used without over pressuring the SEC system. Considering that most polymers synthesised in industry and academia are analysable by SEC using THF as a solvent, it is no surprise that most groups only possess SEC systems running on THF. Under such circumstances, P(HEA) polymers can still be analysed provided the hydroxyl group on the monomer is protected before polymerization (for example with trimethylsilyl groups). After characterisation, deprotection can be achieved easily using a diluted acid solution.19 Other commonly used techniques for obtaining information on mass and PDI for P(HEA) polymers are MALDI-TOF and ESI (negative mode).When analysis of P(HEA) based block copolymers is not possible by SEC, or if additional evidence of block copolymerization is required, gradient polymer elution chromatography (GPEC)23 is an important and useful technique. In GPEC, the sample is introduced into a system pumping a poor solvent for the polymer causing the polymer to precipitate on the column. A gradient is then run in which the composition of a good solvent for the polymer is gradually increased, thus resulting in polymer dissolution when an appropriate solvent composition is reached. The retention time is influenced by the chemical character and solubility (thus mass to some extent) of the polymer.23
Homopolymerization of HEA/HEMA by living radical polymerization (LRP)
Living polymerization was introduced by Szwarc in 1956.24 The most interesting characteristics of this polymerization are that termination does not occur and if polymerization is allowed to occur until monomer consumption is complete, adding a second monomer results in continuation of chain growth since the polymer chains never ‘die’. Thus, the construction of block copolymersvia sequential monomer addition is possible25 and complex macromolecular architectures can be obtained due to increased levels of control over the synthesis. Though efficient in producing well-controlled polymers, living polymerization based on either ionic polymerizations or polymerizations through coordination chemistry suffers from specific drawbacks; (i) extreme sensitivity to impurities (ii) stringent reaction conditions and (iii) the polymerization of functional monomers with hydroxyl groups (e.g. HEA/HEMA) is not possible unless they are protected.2 Living radical polymerization (LRP) techniques, first introduced by Otsu26 who investigated the use of iniferters to control radical polymerization, on the other hand, are more tolerant to impurities and a wide range of functionalities. This section highlights studies of the homopolymerization of HEA/HEMA by LRP with focus on articles in which the polymerization was first demonstrated.Atom transfer radical polymerization (ATRP)
Discovered independently by Sawamoto27 and Matyjaszewski28 at the same time, ATRP is the most widely studied among the LRP techniques. This process takes place in the presence of an alkyl halide initiator and a catalyst formed from a transition metal complexed by a ligand.2Polymerization of HEA was first reported to work using ATRP by Coca et al.29 who demonstrated HEA could be polymerized in a controlled fashion in bulk and in solution using water as the solvent. From their kinetics studies, the polymerizations were found to exhibit first order kinetics and molecular weights were observed to increase with conversion indicating the livingness of the system. Low PDIs were achieved for P(HEA) polymers with Mn of up to 78 000 g/mol but higher PDIs were observed for Mn close to 100 000 g/mol suggesting side reactions take place for high Mn polymers. A year later, the same group also reported the polymerization of HEMA by ATRP.19 The polymerization was initially attempted in bulk at 90 °C but this gave polymers with broad PDIs (1.5). Better control was achieved by polymerizing at 50 °C and 70 °C using a copper chloride catalyst in the presence of a in a mixed polar solvent (methyl ethyl ketone/propanol) to cater for monomer polarity. PDIs of less than 1.5 were achieved below 80% conversion with Mn values below 40 000 g/mol. It was suggested in this article that higher molecular weights are achievable by polymerizing HEMA with the hydroxyl group protected using a trimethylsilyl group (HEMA-TMS). Using this approach P(HEMA-TMS) polymers with Mn of up to 100 000 g/mol were prepared with PDIs below 1.5. P(HEMA) was then obtained by deprotection of the hydroxyl groups using an acid. Armes and co-workers30 went on to demonstrate that the polymerization of HEMA by ATRP can be achieved at room temperature in methanol or water/methanol mixtures. Using CuBr or CuCl catalysts, polymers of Mn less than 10 600 g/mol with low PDIs 1.2–1.3 were obtained suggesting the polymerizations were well controlled.Vargün and Usanmaz17 have also published a study on polymerization of HEA by ATRP. Their polymerizations were conducted both in bulk and in solution. However, no SEC data was supplied in this work.
Nitroxide mediated polymerization (NMP)
In NMP, the polymerization is carried out in the presence of a stable radical such as 2,2,6,6-tetramethyl-piperidin-1-oxyl (TEMPO) which is capable of undergoing reversible termination reactions with active radicals from propagating polymer chains. The trapping of propagating radicals by TEMPO to form a dormant species minimises the amount of active radicals in the system and thus the chances of irreversible termination between propagating chains are lowered.31 In 2005, Bian and Cunningham18 reported the controlled polymerization of HEA by NMP. Their polymerizations were undertaken in bulk and in solution using DMF or water as solvents. Generally, good control was achieved for polymers with PDIs less than 1.3 being observed in their polymerizations for Mn below 100 000 g/mol. At higher Mn however, high PDIs (>1.6) were observed which were attributed to chain branching caused by chain transfer. In addition to the kinetics studies confirming the livingness of their system, block extension of the P(HEA) polymers with butyl acrylate was successfully accomplished.Reversible addition fragmentation chain transfer (RAFT) polymerization
RAFT polymerization was reported simultaneously in 1998 by Rizzardo and co-workers32 at CSIRO (Australia) and by Zard and coworkers in collaboration with Rhodia (France).33 Unlike ATRP and NMP in which polymerization is controlled via reversible termination, RAFT/MADIX polymerization relies on the degenerative chain transfer of a chain transfer agent (CTA), among which thiocarbonyl thio compounds are the most effective.34–38Lai and co-workers39 first reported the synthesis of P(HEA) by RAFT polymerization using a trithiocarbonate CTA. The polymerization was undertaken in solution and low molecular weight P(HEA) with PDI of 1.1 was obtained. However, no kinetics data were supplied in this article. A more detailed study showing kinetics data and chain extension experiments was published by Zhang et al.20 who undertook solution polymerization of HEA in the presence of a trithiocarbonate CTA. Polymers with PDIs of 1.3–1.4 were obtained. The RAFT polymerization of HEMA was reported by Rizzardo and co-workers32 who made an ABA triblock copolymer of MMA and HEMA with MMA as the middle block. Low PDI of 1.2 was observed indicating the polymerization was well controlled.
Materials
Polymeric micelles/nanoparticles
Micelles/polymeric nanoparticles have attracted a lot of interest due to their potential as drug delivery agents.11,40,41 Polymeric nanoparticles are often derived from amphiphilic block copolymers which comprise of a hydrophilic block and a hydrophobic block. Such copolymers are capable of self assembling into core shell structures such as micelles, vesicles, rods above their critical micellization concentration (CMC).42 Due to the hydrophilic character of P(HEA) and P(HEMA) (low Mn), these polymers have been used as hydrophilic blocks in designing polymeric micelles/nanoparticles.Zhang et al.20 synthesised a range of well-defined P(HEA)-b-P(BA) amphiphilic block copolymersviaRAFT polymerization with different block lengths to produce core cross-linked micelles. Self assembly of these copolymers was achieved by dissolving P(HEA)-b-P(BA) in methanol which is a good solvent for both blocks followed by gradual addition of an excess of water which is a good solvent for P(HEA) but not for P(BA) resulting in the formation of micelles. The size of micelles was found to be dependent on the block lengths. Stabilisation of the micellar aggregates was achieved through core cross-linking viaRAFT polymerization applying hexanediol diacrylate as a cross-linker yielding the desired nanostructures (Fig. 2).
 | ||
| Fig. 2 Preparation of core cross-linked micelles (adapted from ref. 20). | ||
Bulmus and co-workers43 also reported the synthesis of P(HEA)-b-P(BA) copolymers using RAFT polymerization which were used to generate acid-labile core cross-linked micelles for pH-triggered release of antitumor drugs. In this work, block copolymers with PDIs of 1.1–1.4 were synthesised using 4-cyanopentanoic acid dithiobenzoate (CPDB) as the CTA in ethanol. Micellisation was induced by addition of aqueous phosphate buffer (pH = 8.5) containing 2,2′-Azobis[2-(2-imidazolin-2-yl) propane] dihydrochloride initiator to an ethanol solution of the copolymer, doxorubicin and the acid labile di(2-acryloyloxy ethoxy)-[4-hydroxyphenyl] methane cross-linker. Polymerization of the diacrylate cross-linker by RAFT resulted in the formation of core cross-linked micelles with diameters of 50–180 nm. Loading capacities of up to 60% (w/w) were obtained which indicates these micelles are good carriers of hydrophobic drugs. The pH-triggered release of the drug was assessed by UV-vis at pH 5 (cancerous cells have low pH) and at pH 7.4 (typical for normal cells). It was observed that doxorubicin release was faster at pH 5 than at pH 7.4 indicating these micelles offer site-selective drug release which is ideal for drug delivery applications.
The preparation of pH-responsive shell cross-linked micelles which act as nanoreactors for preparation of gold nanoparticles has been reported by Armes' group.44P(EO)-b-P(HEMA)-b-P(DEAEMA) was synthesised by ATRP starting from a P(EO) macroinitiator and the self assembly of these triblock copolymers into micelles was observed to take place at high pH (pH = 7–8) with P(EO) forming the shells whilst P(DEAEMA) formed the core. The intermediary P(HEMA) layer was used to effect cross-linking of the micelles through a reaction between the hydroxyl groups and divinyl sulfone to yield cross-linked micelles which were found to have a pH dependant swelling behaviour. Treatment of the cross-linked micelles with HAuCl4 resulted in protonation of P(DEAEMA) which became electrostatically bound to AuCl4− ions and addition of sodium borohydride reduced AuCl4− to Au thus producing gold containing nanoparticles (Fig. 3). These nanoparticles were observed by TEM and diameters of about 45 nm.
 | ||
| Fig. 3 Preparation of gold loaded nanoparticles (adapted from ref. 44). | ||
Thermoresponsive block copolymers containing P(NiPAAM) as the hydrophobic block (above LCST) and P(HEA) as the hydrophilic block have also been reported. Lu and co-workers45 made P(NiPAAM)-b-P(HEA) copolymers by RAFT polymerization which were found to form micelles in water upon temperature change to above the LCST of P(NiPAAM).
In a recent publication, Zhuo and co-workers46 demonstrated the design of multicolored micellar complexes using P(NiPAAM-co-HEMA)-b-P(VP) block copolymers. Using RAFT polymerization, P(NiPAAM-co-HEMA)-b-P(VP) copolymers were synthesised and subsequently conjugated to fluorescein isothiocyanate to yield fluorescent amphiphilic copolymers. P(NiPAAM) has an LCST of 32 °C in water above which it loses its hydrophilic character. However, it is well known that the copolymerization of hydrophilic monomers with NiPAAM increases its LCST significantly. Thus, within the temperature range of their LCST determination (10–60 °C), P(NiPAAM-co-HEMA) had no LCST indicating the polymer was hydrophilic due to HEMA incorporation. Self assembly of the fluorescent copolymers in water resulted in spherical micelles which were about 50 nm in diameter. The fluorescence of the micellar complexes was found to be temperature and pH dependent thus the authors proposed these complexes may find use in pH/temperature probing and bioanalysis.
Cao et al.47 prepared 3 arm P(HEMA)-b-P(NiPAAM) star polymers by ATRP with DPs 40 and 320/160 for HEMA and NiPAAM, respectively. Using a tri-arm ATRP initiator, HEMA was polymerized to give a well defined star P(HEMA) polymer with low PDI (1.25) which was then chain extended with NiPAAM to give the thermoresponsive block copolymer. The authors however did not report the PDI after block extension. P[(HEMA)40-b-P(NiPAAM)320]3 was found to form micelles in water with diameters of about 100 nm.
Holder and co-workers48 obtained micelles from amphiphilic triblock copolymers of P(HEMA) and poly(methylphenylsilane), P(MPS), in water which were made by ATRP. The DP of HEMA segments was kept low to ensure the P(HEMA) was hydrophilic and had water solubility. They synthesised an ω, α-dihalo P(MPS) macroinitiators which were used to polymerize HEMAviaATRP resulting in P(HEMA)-b-P(MPS)-b-P(HEMA) triblocks. Though the PDIs of their final products were high (PDI = 1.5–1.9), the polymerization of HEA seems to have been well controlled taking into account the PDIs of their macroinitiators were high to begin with (PDI = 1.6–1.95).
Armes and co-workers4 have successfully synthesised amphiphilic P(HEMA)-b-P(MMA) by ATRP which formed micelles of about 17 nm in diameter in acidic water (pH = 2.5). The success of micelle formation was confirmed by 1H NMR which showed the disappearance of the hydrophobic P(MMA) signals from the spectrum upon dilution of the block copolymer solution in deuterated methanol with D2O/DCl. The disappearance of the signals was attributed to the fact that the P(MMA) formed the micelle cores and thus became “hidden”.
Using RAFT polymerization, Grignard et al.49 synthesised fluorinated poly(1H,1H,2H,2H-heptadecafluorodecyl acrylate)-b-P(HEA) block copolymers which formed spherical micelles upon slow dissolution in ethanol. Micelles with diameters of 50–100 nm were observed.
Amphiphilic ethyl cellulose (EC)-graft-P(HEMA) copolymers have been reported.50 EC was modified into an ATRP initiator which was used to initiate the polymerization of HEMA resulting in EC-g-P(HEMA) copolymers with PDIs mostly below 1.2 indicating the polymerizations were well controlled. The graft polymers dissolved molecularly in DMF and when dialysed against water, spherical micelles with P(HEMA) coronas and EC cores having diameters of about 50–100 nm were formed.
Schmidt and co-workers51 designed protein (Annexin-A5) decorated nanoparticles starting from P(St)-b-P(HEMA) made by ATRP. To obtain nanoparticles, the block copolymer was subjected to partial phosphorylation, silylation and methanolysis of the hydroxyl groups to yield P(St)-b-P(2-phosphatethyl methacrylate-co-HEMA) (P(St)-b-(PEMA-co-HEMA)) which formed spherical micelles in an aqueous buffer at pH 8. Through light scattering studies, Annexin A5 was observed to bind to the micelles through the hydrophilic P(PEMA-co-HEMA) blocks resulting in the formation of biofunctionalised nanoparticles. These nanoparticles were reported to have potential applications in areas of in vivo imaging, targeted drug delivery and biochips.
Our group reported the synthesis of poly(ethyl acrylate-b-(HEA-co-N-acryloxysuccinimide) viaRAFT polymerization and their self assembly in water to form spherical micelles of diameter ca. 40–60 nm.52 Reaction of N-acryloxysuccinimide with a diamine led to the crosslinking of the hydrophilic shell to yield soft nanoparticles. The polymer synthesis strategy we used ensured the positioning of the thiocarbonyl thio group mediating RAFT polymerization on the surface of the particles shell. The reduction of this group into thiols yielded thiol-decorated nanoparticles, which could be functionalized using thiol-ene click chemistry under mild conditions, to form nanoparticle-based gels, fluorescent-tagged particles, and protein-nanoparticle conjugates.
Where CTAs with long hydrophobic chains are used in RAFT polymerization to make block copolymers, hydrophobic end groups can affect the amphiphilic character of the resultant block if it's on the hydrophilic end. In such cases, end group modification to maintain polarity of the block may be desirable.
Complex polymeric architectures and their corresponding nanostructures
Complex polymeric architectures such as star, comb and (hyper)branched polymers have attracted a lot of interest as they tend to have different properties from the linear counterparts.53 For example, for a given polymer, the star shaped polymer tends to have a more compact structure with high functionality and low viscosity when compared to the linear polymer of the same molecular weight.54 Thus such polymers have possible processing advantages over linear polymers.55Design of complex architectures using hydroxyl groups as initiators for ring-opening polymerization (ROP)
The ability of the hydroxyl group to initiate ROP of cyclic esters allows easy access to complex polymeric architectures starting from small molecules containing multiple hydroxyl functionalities as well as polyols.The synthesis of brush block copolymers with P(CL)-b-P(BA) side chains was reported by Lee and co-workers.56 Herein, P(HEMA) with PDI of 1.2 was synthesised viaATRP followed by polymerization of ε-CL initiated by the P(HEMA) hydroxyl groups using tin(II)2-ethyl hexanoate as the catalyst to yield P(HEMA)-g-P(CL). Following the modification of the hydroxyl end groups on the P(CL) grafts into ATRP initiators using 2-bromopropionyl bromide, polymerization of BA was undertaken to obtain well defined P(HEMA)-g-P(CL)-b-P(BA) copolymers (PDI < 1.2). The core-shell structure of the brushes was confirmed by AFM. In the same year, the synthesis of P(HEMA)-g-P(CL) copolymers using the same approach was also reported by the same group.57 A few years later, this group extended their synthetic approach to amphiphilic brush-block-brush copolymers wherein a multifunctional initiator, P(BiBEM)-b-P(HEMA), was synthesised by ATRP and used for ROP of ε-CL followed by ATRP of BA through P(BiBEM) units yielding [P(BiBEM)-g-P(BA)]-b-[P(HEMA)-g-P(CL)].58 These brush-block-brush copolymers were observed to form flower-like and dumbbell-like structures due to their amphiphilic nature (Fig. 4).
![Synthesis of [P(BiBEM)-g-P(BA)]-b-[P(HEMA)-g-P(CL)] brush-block-brush copolymers (adapted from ref. 58).](/image/article/2011/PY/c0py00160k/c0py00160k-f4.gif) | ||
| Fig. 4 Synthesis of [P(BiBEM)-g-P(BA)]-b-[P(HEMA)-g-P(CL)] brush-block-brush copolymers (adapted from ref. 58). | ||
The synthesis of P(MMA-co-HEMA)-g-P(CL) copolymersviaATRP and enzyme catalysed ROP in supercritical carbon dioxide was reported by Howdle and co-workers59 using two approaches. In their first approach, P(MMA-co-HEMA) was synthesised by ATRP followed by enzymatic ROP of ε-CL to obtain graft copolymers with relatively high PDIs (∼2.0). Their second approach involved a one pot reaction in which all monomers, ATRP catalysts and the enzyme were mixed together and reacted to have ATRP and ROP taking place simultaneously. This second approach yielded graft copolymers which had lower PDIs (1.4–1.7) than observed in the first approach suggesting better control was achieved via the one pot approach. An advantage of their synthetic approach is that the graft polymers produced contain unreacted free hydroxyl groups which allow for further functionalisation and also the construction of more complex architectures. However, low grafting densities were observed which was attributed to steric hindrance. Studies on improving the grafting densities were later published by the same group.60 It was observed that improving the randomisation of HEMA in P(MMA-co-HEMA) or having the hydroxyl groups further away from the backbone by using a longer molecule results in improved grafting densities (80%–100% grafting). These polymers were reported to be potentially suitable for biomedical applications.
Xu and Huang61 reported the preparation of P(HEMA-co-St)-g-P(CL)viaRAFT polymerization and ROP. In this study, P(HEMA-co-St) was synthesised viaRAFT polymerization in the presence of a dithiobenzoate CTA which gave copolymers with PDIs of 1.1–1.4. Using these copolymers as initiators, ROP of ε-CL was undertaken with stannous octoate as the catalyst and reasonably well defined P(HEMA-co-St)-g-P(CL) graft polymers with PDIs under 1.4 were obtained. The same authors went on to report the report the synthesis of amphiphilic [P(EO)-b-P(HEMA-co-St)]-g-P(CL) using a similar approach to the one described previously.62 For this work, P(EO) was modified into a CTA via an esterification reaction and the resulting macroCTA was used to mediate the copolymerization of HEMA and styrene affording P(EO)-b-P(HEMA-co-St) which was used to initiate the ROP of ε-CL. Later, the same group broadened the scope of their synthetic approach to more complex brush-type amphiphilic block copolymers.63 Similar to the previous study, PEO was modified into a macroCTA via esterification but the PEO used in this case was terminated with hydroxyl groups on both chain ends resulting in a bifunctional macroCTA being formed. The copolymerization of HEMA and styrene in the presence of this macroCTA thus gave P(HEMA-co-St)-b-P(EO)-b-P(HEMA-co-St) which was employed to initiate the ROP of ε-CL to yield [P(HEMA-co-St)-g-P(CL)]-b-P(EO)-b-[P(HEMA-co-St)-g-P(CL)] copolymers with narrow MWD (PDIs < 1.5). The copolymers formed vesicles, rods or pearl necklace morphology in THF/water solutions depending on the amount of water present in the system.
Wu et al.64 have reported the polymerization of HEMA by ATRP which was used for the synthesis of mixed PLLA/PSt molecular brushes. P(HEMA) was partially esterified using 2-bromopropionyl bromide to yield P(BiBEM)-co-P(HEMA) copolymers which were used to initiate ROP of L-lactide and AGET ATRP of styrene. The tin(II) 2-ethylhexanoate which was used to catalyse the ROP reaction also served as the reducing agent for the AGET ATRP reaction allowing the reactions to be carried out simultaneously. The resulting P(BiBEM)-g-P(St)-co-P(HEMA)-g-PLLA copolymers generally had PDIs ≤ 1.6 suggesting some degree of control existed though it was not ideal. Due to immiscibility of PLLA and P(St) chains, the brush copolymers were observed to undergo phase separation forming star-like nanostructures when cast into thin films on mica. Annealing of the thin films at high temperatures led to the formation of globular nanostructures.
Amphiphilic comb-block-comb copolymer brushes were reported by Zhao and co-workers65 who synthesised [P(HEMA)-g-PLLA]-b-Poly(ethylene glycol monomethyl ether methacrylate) ([P(HEMA)-g-PLLA]-b-P(EOMA) copolymers by ATRP and ROP. P(HEMA) was synthesised by ATRP followed by the simultaneous polymerization of LLA by ROP and EOMA macromonomer by AGET ATRP in the presence of tin(II)2-ethyl hexanoate as the catalyst/reducing agent (vide supra). High Mn were achieved (125 000 g/mol–222 000 g/mol) and low PDIs were observed (1.1–1.3) indicating the polymerizations were well controlled. The comb-block-comb copolymer brushes were observed by AFM and their self assembly in water led to micelle formation which was established by TEM. This group has a strong interest in the area of comb copolymers syntheses and they have also published an in-depth study on crystallisation and melting behaviour of PLLA comb copolymers.66 The study was aimed at probing the physical properties of these materials which are an important factor when looking at potential applications.
Mecerreyes et al.67 have also reported the preparation of graft copolymers by ROP of ε-CL and ATRP of MMA and HEMA in a single step to generate P(MMA-co-HEMA)-g-P(CL) copolymers. Analysis of the polymers by 1H NMR and SEC studies before and after PCL hydrolysis proved graft polymers had been formed. This approach is deemed to be attractive for production of biodegradable graft copolymers in the industry due to the simplicity of the procedure. This approach was later extended to the synthesis of branched polymers by the same group wherein ε-CL modified into an ATRP initiator was reacted with HEMA in the presence of an ROP catalyst.68 The modified ε-CL was capable of initiating the ATRP of HEMA as well as undergoing ROP whilst HEMA could undergo polymerization and initiate ROP of modified ε-CL through hydroxyl groups. Thus, adducts of the reactions produced multiple reaction sites through which further reactions could take place producing branched polymer structures (Fig. 5). The simultaneous polymerization of ε-CL, modified ε-CL, MMA and HEMA was also studied with the purpose of modifying the branching density. Evidence of branched polymer formation was obtained viaSEC analysis which showed traces with multimodal distributions and broad PDIs which are accepted for branched polymers.

Zheng and Stover69 have reported the preparation of microspheres grafted with P(CL)-b-P(DMAEMA). The microspheres were derived from lightly cross-linked P(DVB-co-HEMA) which was used to initiate the ROP of ε-CL with different catalysts (triethylaluminium or aluminium isopropoxide) followed by end group modification of the P(CL) chains into ATRP initiators. Block copolymerization was then achieved by ATRP of DMAEMA to give microsphere-g-P(CL)-b-P(DMAEMA) which were observed by ESEM and TEM. The grafting of P(CL) was observed to depend on the ROP catalyst used and DMAEMA grafting of more than 70% was achieved.
Graft copolymers have also been synthesised by Ydens et al.70 who copolymerized HEMA and MMA by ATRP to produce ROP initiators which were subsequently used for the polymerization of L-lactide or ε-CL using stannous octoate as a catalyst.
Star shaped amphiphilic copolymers have been synthesised by ATRP and ROP as reported by Jia et al.71P(HEMA)-g-P(CL)-b-P(DMAEMA) and P(HEMA)-g-P(CL)-b-P(t-BMA) star shaped polymers were prepared by ROP of ε-CL using hyperbranched P(HEMA) as initiators followed by ATRP of either DMAEMA or t-butyl methacrylate (t-BMA) after conversion of P(HEMA)-g-P(CL) into an ATRP initiator. Where t-butyl methacrylate was polymerized, the block copolymer was subjected to hydrolysis to obtain P(HEMA)-g-P(CL)-b-P(MAA) with poly(methacrylic acid) (P(MAA)) as the hydrophilic block. The self assembly of these polymers in water resulted in the development of spherical micelles with average diameters of 120 nm–210 nm based on TEM studies.
The production of graft polymersvia simultaneous RAFT and ROP was reported by Barner-Kowollik and co-workers.72 In this work, HEMA and ε-CL were polymerized in the presence of stannous octoate and a dithiobenzoate CTA to form P(HEMA)-g-P(CL) in a single step. Grafting densities greater than 90% were achieved and PDIs were mostly in the range 1.3–1.7. The high PDIs were attributed to bimolecular termination and transesterification of the P(CL) chains.
Our group demonstrated that RAFT polymerization and bifunctional sparteine/thiourea organocatalyst-mediated ROP could be combined to produce poly(L-lactide) star polymers and poly(L-lactide-co-styrene) miktoarm star copolymers architecture following a facile experimental procedure, and without the need for specialist equipment.73 RAFT was used to copolymerize ethyl acrylate (EA) and HEA into poly(EA-co-HEA) co-oligomers of degree of polymerization 10 with 2, 3, and 4 units of HEA, which were in turn used as multifunctional initiators for the ROP of L-lactide, using a bifunctional thiourea organocatalytic system. Furthermore, taking advantage of the living nature of RAFT polymerization, the multifunctional initiators were chain extended with styrene (poly((EA-co-HEA)-b-styrene) copolymers), and used as initiators for the ROP of L-lactide, to yield miktoarm star copolymers.
Complex polymeric architectures and their nanostructures through postmodification of P(HEA)/P(HEMA)
As discussed above, the hydroxyl group is capable of undergoing a wide variety of reactions, thus postmodification of hydroxylated polymers is possible and offers a route to designing complex polymeric architectures.Recently, Chun and co-workers have synthesised graft copolymersviaSET polymerization and ATRP starting from P(HEA) based block copolymers. P(NiPAAM)-b-P(HEA) copolymers made by SET polymerization were converted into ATRP initiatorsvia a reaction between the P(HEA) hydroxyl groups and 2-chloropropionyl chloride followed by the ATRP of the desired monomer (MX) to obtain P(NiPAAM)-b-[P(EA)-g-P(MX)] copolymers. Using this approach, DMAEA was polymerized to give double hydrophilic graft copolymers which were used to produce polymer stabilised gold nanoparticles.74 The nanoparticles were observed by TEM (4 nm–30 nm diameter) and the stability was assessed by zeta potential. In another publication,75 the same group polymerized DMAEMA which is to obtain double hydrophilic graft copolymers with pH (DMAEMA) and thermoresponsive (NiPAAM) characteristics. The self assembly of these copolymers in water was then studied at various pH and temperatures. Vesicles were observed at high pH/room temperature whilst spherical micelles with P(NiPAAM) cores and positively charged P(DMAEMA) coronas were formed above the LCST of P(NiPAAM) (32 °C) at low pH. Most recently, they have also published similar work with the pH-responsive monomer being poly(2-vinylpyridine).76 The micellisation of the resulting copolymers was also studied as before. The same group has also used a similar approach to prepare amphiphilic copolymers graft copolymers based on ferrocene.77 In this instance, α-bromoisobutyryl bromide was used to modifify the P(HEA) hydroxyl groups into initiators and 2-acryloyoxy-ethyl ferrocenecarboxylate (AEFC) was then polymerized by ATRP. The electro-activity of the P(NiPAAM)-b-[P(EA)-g-P(AEFC)] copolymers was ascertained by cyclic voltammetry studies. Self assembly studies revealed the polymers formed micelles or vesicles depending on the length of P(AEFC) and the aggregates sizes were affected by the preparation method. Since the aggregates had redox properties, it was suggested that these materials can potentially be used as redox drug-controlled release carriers.
Polymers with dumbbell shape were reported by Dimtrov et al.78 who synthesised high molecular weight P(BA-co-tBA) (Mn > 100 000 g/mol) using a bifunctional ATRP initiator followed by chain extension of the polymer with HEA to obtain P(HEA)-b- P(BA-co-tBA)-b-P(HEA). The copolymer was treated with α-bromoisobutyryl bromide which reacted with the hydroxyl groups to obtain multiple ATRP initiation sites which were used to initiate the ATRP of BA/tBA affording the dumbbell shaped polymers (Fig. 6). The dumbbell polymers were characterised by SEC and SEC-MALLS which showed molecular weights of up to 510 000 g/mol were achieved and they were well defined with PDIs less than 1.4 being observed.
 | ||
| Fig. 6 Synthesis of dumbell shaped P(BA) polymers (0.9 eq. BA + 0.1 eq. t-BA refers to monomer(s) to initiator ratio; adapted from ref. 78). | ||
Comb-like thermoresponsive copolymers prepared viaRAFT polymerization and ATRP have been reported by Wan et al.79RAFT polymerization was used to prepare P(ACN)-b-P(HEMA) and P(ACN)-co-P(HEMA) copolymers which were modified into an ATRP polyinitiator by reacting the hydroxyl groups of HEMA with 2-bromoisobutyryl bromide followed by the ATRP of P(NIPAAM) with these initiators. Good control was achieved for these polymerizations with the PDIs for both P(ACN)-b-[P(HEMA)-g-P(NiPAAM)] and P(ACN)-co-[P(HEMA)-g-P(NiPAAM)] being about 1.4. The contact angle of water on thin copolymer films was measured various temperatures and it was observed that the surfaces had temperature dependant wettability which was influenced by the copolymer shape and content of NiPAAM. These polymers were reported to be potentially applicable in making functional fibers or membranes.
Matyjaszewski's group reported the preparation of densely grafted copolymers by ATRP wherein TMS protected HEMA was polymerized by ATRP followed by TMS hydrolysis to obtain P(HEMA) which was modified into an ATRP macroinitiator via an esterification reaction with 2-bromoisobutyryl bromide.80 The ATRP macroinitiator was used to initiate styrene and BA polymerizations at high dilution to reduce termination reactions. The graft polymers were visualised by AFM and also analysed by triple detector SEC which showed the polymerization were well controlled as evidenced by the low PDIs (<1.4). This group has also reported the synthesis of macromolecular bottle-brush copolymers using a similar approach.81 Following the approach used by the Matyjaszewski's group,80 Cheng et al.82 reported the synthesis of amphiphilic core-shell brushes by ATRP in which the ATRP macroinitiator derived from P(HEMA) was used to initiate styrene or tBA polymerization followed by chain extension of the graft polymer with tBA or styrene. The resulting graft block copolymers with P(St)-b-P(tBA) or P(tBA)-b-P(St) side chains then underwent hydrolysis to convert P(tBA) into poly(acrylic acid). Analysis of the copolymers by AFM before hydrolysis revealed the macromolecules as worm-like nanostructures whilst the hydrolysed copolymers formed wormlike cylindrical micelles.
Recently, Gao and Matyjaszewski83 prepared molecular brushes using ATRP and copper catalysed ‘click’ reactions. In this work, P(HEMA) was synthesised by ATRP then subjected to esterification conditions in the presence pentynoic acid yielding P(HEMA) with pendant alkyne groups. Azide terminated P(St), P(EO), P(BA) and P(BA)-b-P(St) prepared by ATRP were then ‘clicked’ onto the alkyne functionalised P(HEMA) producing a wide range of grafted polymers. The grafting densities in this case were however low, generally less than 50%, and this was attributed to the reduced accessibility of the reactive sites through steric hindrance as the ‘click’ reactions progressed.
Khelfallah et al.84 reported the synthesis and self assembly of amphiphilic brush-coil block graft copolymers. Herein, P(St)-b-P(HEMA) synthesised by ATRP was modified into an ATRP macroinitiator through the P(HEMA) hydroxyl groups followed by the polymerization of tBA by ATRP. Acid hydrolysis gave the amphiphilic P(St)-b-P(EMA)-g-P(AA). Star-like micelles were observed from the self assembly studies.
Y-shaped acidic polymers were reported by the Armes group.85 They converted amine capped poly(alkylene oxides) (Jeffamine) into bifunctional ATRP initiators and then polymerized HEMA to obtain (Jeffamine)-b-P(HEMA)2 which was subsequently reacted with succinic anhydride to esterify the P(HEMA) blocks. The P(HEMA) was transformed into acidic poly(2-(succinyloxy)ethyl methacrylate) (P(SEMA)) thus forming Y-shaped Jeffamine-b- P(SEMA)2.
Armes and co-workers86 have also reported the synthesis of well defined polyacids starting from P(HEMA) prepared by ATRP. The hydroxylated polymers were reacted with either succinic anhydride or 1,2-cyclohexanedicarboxylic acid anhydride in the presence of a base to produce P(SEMA) and poly(2-(2-(carboxylic acid)cyclohexylcarboxy)ethyl methacrylate) (P(CEMA)). Studies to determine and optimise the esterification process were undertaken and degrees of esterification of up to 99% could be achieved indicating the process was indeed an efficient way of obtaining polyacids. Moreover the products were well defined with PDIs below 1.4. P(EO)-b-P(SEMA) was also synthesised and the micellisation behaviour of this polymer was studies at various pH values taking advantage of the pH-responsiveness of the P(SEMA) block. DLS and 1H NMR studies indicated that aggregates were formed at pH 3 where P(SEMA) was water insoluble. The same group87 has used a similar approach to make zwitterionic block copolymers from block copolymers of a range of tertiary amine methacrylates (TAM) with HEMA. The TAM used were DMAEMA, DEAEMA, DPAEMA and MEMA. Herein, P(TAM)-b-P(HEMA) was reacted with succinic anhydride to convert the P(HEMA) block into P(SEMA) thus giving a diblock in which both blocks exhibit pH-responsiveness. The diblocks formed colloidal aggregates in water at low pH 2 and at high pH (above pKa of P(TAM)) as observed from 1H NMR and DLS measurements (Fig. 7). This approach was also extended to the design of pH-responsive P(EO)-b-P(DMAEMA)-b-P(SEMA) triblock copolymers which were observed to form three types of colloidal aggregates depending on the pH.88 Through detailed 1H NMR and DLS studies it was concluded that the aggregate formation was driven by hydrogen bonding, interpolyelectrolyte complexation and hydrophobic interactions. The same group has also reported the synthesis of pH-responsive Y-shaped zwitterionic P(DMAEMA)-b-P(SEMA)2 block copolymers89 starting from P(DMAEMA) with two ATRP initiation sites which was used to polymerize HEMA followed by esterification as described previously.85 The micellisation behaviour of these zwitterionic polymers was also studied at various pH values and aggregation formation was observed at low and high pH values similar to the P(DMAEMA)-b-P(SEMA) diblocks they reported earlier.87
 | ||
| Fig. 7 Preparation of zwitterionic polymers and their aggregation behaviour at different pH values (adapted from ref. 87). | ||
Lately, Armes and co-workers prepared well defined strong acid homopolymers and diblock copolymers by ATRP starting from hydroxylated polymers.90 The esterification of hydroxylated homopolymers/block copolymers using 2-sulfobenzoic acid cyclic anhydride (SBA) was employed to obtain strong polyacids with very high degrees of esterification being realised. The simplicity of their approach makes it an attractive way producing strongly acidic homopolymers/block copolymers for a wide variety of applications.90 In a recent publication,91 this approach was used to synthesise amphiphilic diblock copolymers starting from P(BA)-b-P(HEMA) and P(BA)-b-P(HEA) made by ATRP which were modified into polyacids using SBA. The resulting amphiphilic copolymers were observed to form spherical micelles, cylinders and cylindrical aggregates. The application of these block copolymers as stabilisers for the emulsion polymerization of BA were demonstrated herein.
The use of RAFT polymerization to prepare phosphonated polymers and their corresponding polyacids through P(HEA) modification has been reported.92 These polymers were synthesised from random and block copolymers containing vinylidine chloride, MA, and HEA were made by RAFT polymerization and modified into phosphonated copolymers through a reaction between hydroxyl groups of P(HEA) units and phosphonated epoxide. A complete substitution of the hydroxyl groups was achieved and upon hydrolysis of the phosphonated copolymers, the phosphonate groups were converted into either monoacid or diacid groups depending on the conditions employed.
Yusa et al.93 reported the preparation of photo-cross-linked micelles from P(EG)-b-P(DEAEMA-co-HEA) copolymer synthesised by RAFT polymerization which was modified into P(EG)-b-P(DEAEMA-co-CEA) using cinnamoyl chloride to provide cross-linking sites. These copolymers formed micelles in aqueous solution above pH 7 with P(DEAEMA-co-CEA) cores and micelle cross-linking was accomplished by UV initiated photodimerisation of the cinnamoyl groups. The cross-linked nanostructures were observed by TEM and their ability to capture and release hydrophobic molecules depending on pH was studied. Hydrophobic molecules were captured at high pH where P(DEAEMA) core is hydrophobic whilst release occurred at low pH where the polymer is hydrophilic. This approach has also been used to make intermediary layer cross-linked micelles starting from P(EG)-b-P(HEMA)-b-P(MMA) copolymers which were converted to P(EG)-b-P(CEA)-b-P(MMA) placing the cross-linking sites between the hydrophilic and hydrophobic blocks.94 Spherical nanoparticles were observed by TEM after self assembly and cross-linking.
Chang and Pugh prepared comb polymers of P[11-4(4′-cyanophenyl-4′′-phenoxy)undecyl acrylate]viaATRP. An ATRP macroinitiator was first synthesised by copolymerizing 11-4(4′-cyanophenyl-4′′-phenoxy)undecyl acrylate with protected HEA which was subsequently deprotected by acid hydrolysis then esterified using 2-bromopropionyl chloride. The macroinitiators were then used to polymerize 11-4(4′-cyanophenyl-4′′-phenoxy) undecyl acrylate affording the comb polymers.95
Durmaz et al.96 reported the preparation of graft copolymers by a combination of ATRP and a photochemical acylation process. P(MMA-co-HEMA) and P(St) were synthesized by ATRP followed by end group modification of the P(St) polymer with benzodioxinones. The benzodioxinone functionalized P(St) polymers were reacted with in the presence of benzophenone under UV light (300 nm) and P(MMA-co-HEMA)-g-P(St) was formed via a reaction between the benzodioxinone and HEMA hydroxyl groups. Moderate grafting efficiencies of 58% and 45% were achieved and this was attributed to steric hindrance and photoactivity of benzophenone released during the photochemical reaction.
Saito and co-workers97 reported the synthesis of ladder like polymersviaATRP of P(2-methacryloyloxyethyl methacrylate) derived from P(HEMA). In this work P(HEMA) was synthesized by ATRP and modified into poly(2-methacryloyloxyethyl methacrylate) via esterification with methacrylic anhydride and the Shotten-Baumann method. Up to approximately 90% conversion of esterification could be realised with both methods and the presence of the vinyl groups in the products was determined by 1H NMR. The ATRP of poly(2-methacryloyloxyethyl methacrylate) made by the Shotten-Baumann method was then conducted at very high dilutions to encourage intramolecular polymerization thus generating ladder like structures. In a separate but related publication, this group has studied the architectural effect of ladder-like polymer on glass transition temperature in which they made polymers with varied the amounts of ladder and studied their thermal properties.98 The same group has also reported a study using similar chemistry in which they investigated the propagation of the polymerization of methacryloyl type multi-vinyl monomers with P(HEMA) backbone.99
Jiang et al.100 have reported the synthesis of degradable P(HEMA) with grafted P(DMAEMA) using ATRP and ‘click’ chemistry. P(HEMA) with pendant alkyne functional groups was first synthesised by reacting P(HEMA) made by ATRP with a propargyl ester of carbonyl diimidazole forming carbonate ester linkages which are degradable (Fig. 8). A copper catalysed ‘click’ reaction between the alkyne functionalised polymer with azide terminated P(DMAEMA) gave the desired graft copolymers. The degradation of the polymers was observed to take place at pH 7.4 and 37 °C (physiological conditions). Application of these polymers in gene delivery was also demonstrated.
 | ||
| Fig. 8 Synthesis of P(HEMA) with pendant degradable linkages (adapted from ref. 100). | ||
Recently, Yang et al.101 reported the preparation of temperature responsive copolymers for DNA condensation and gene transfection studies. Poly(2-(2-methoxyethoxy)ethyl methacrylate)-b-P(HEMA) [P(MEOMA)-b-P(HEMA)] block copolymers were made using ATRP followed by activation of the pendant P(HEMA) hydroxyl groups with 1,1′-carbodiimidazole to obtain reactive groups for the grafting of poly(ethyleneimine) (P(EI)). These graft copolymers exhibited temperature responsiveness and their LCSTs were observed to be below 39 °C. The temperature sensitive DNA condensation and gene transfection by the copolymers was then investigated.
Hybrid (nano)structures
Hybrids are an interesting class of materials because they incorporate structural and functional properties of a synthetic polymer with those of an inorganic or organic naturally occurring material thereby improving the applicability of both materials. For example, conjugation of polymers such as P(EG) to peptide based drugs can result in improved circulation time, water solubility, stability and reduced antigenicity102–104 whilst the grafting of polymers onto metals can simply be to modify the properties of the surface or offer a protective barrier to prevent environmental attack. Thus polymer hybrid materials have been synthesised for a wide range of applications which include drug delivery, bio-sensors, artificial enzymes, photonics, chromatography and nano-electronic devices.105–107 Accordingly, these materials have continued to attract a lot of research interest.Hao and co-workers108 reported the preparation of metal chelating carbon nanotubes which were designed by grafting of P(HEMA) onto the nanotubesviaRAFT polymerization followed by acid hydrolysis of the polymer to give P(MAA). FT-IR and TGA studies proved success of the grafting reaction and the grafted polymer content was found to be 26%–78% depending the polymerization period. The acid functionalised carbon nanotubes were observed to chelate silver ions and the resulting hybrid nanostructures were observed by TEM (Fig. 9). A similar approach has also been reported by Liu et al.109 who prepared metal chelating PVC beads by grafting P(HEA) onto PVC viaATRP followed by hydrolysis of P(HEA) to P(AA). The PVC-P(HEA) and PVC-P(AA) beads were found to scavenge heavy metals (Hg, Zn, Cu and Cd) from aqueous solutions with the amount depending on pH of the solutions. An interesting feature of these beads is that they could be regenerated after each metal chelation cycle making them potentially applicable in column packings for technological applications.
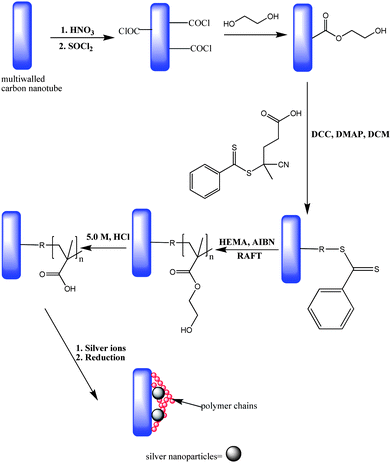 | ||
| Fig. 9 Preparation of carbon natubes-silver metal hybrids (adapted from ref. 108). | ||
Recently, europium coordinating halloysite nanotubes (HNTs) were reported by Wan et al.110 Starting with bromine functionalized HNTs, P(HEMA) and P(MMA)-b-P(HEMA) were grown from the surfaces by ATRP followed by esterification of hydroxyl groups of P(HEMA) with succinic anhydride to obtain HNT-P(SEMA) and HNT-P(MMA)-b-P(SEMA). The hybrids were capable of forming coordination complexes with europium ions in the presence of 1,10-phenanthroline forming photoluminescent hybrids. These materials may be used in fluoroimmunossay or as spectroscopy probes. The polymer grafting was confirmed by TGA whilst metal coordination was observed by FT-IR and fluorescence spectroscopy studies.
Yan and co-workers111 have reported the functionalisation of multiwalled carbon nanotubes (MWNT) by in situ ATRP. MWTN was functionalised with an amphiphilic P(MMA)-b-P(HEMA) layer via surface initiated ATRP. The structure of the product was observed by TEM and the grafting was confirmed to be successful by 1H NMR, FT-IR and TGA. Recently, the same group has used the same approach and reported the grafting of P(MMA)-b-P(HEMA) to titanate nanotubes.112
Preparation of P(HEMA) functionalised P(DVB) microspheres has been reported. In the work reported by Goldmann et al.,113 pendant alkene bonds of cross-linked P(DVB) were reacted with azidoundecane thiol using thiol-ene chemistry to yield azide functionalised microspheres which were then reacted with alkyne terminated P(HEMA)viaCuAAC chemistry. In addition to microscopy and spectroscopic techniques, the grafting was also assessed by reacting the particles with Rhodamine chloride which reacts with hydroxyl groups and, the Rhodamine functionalised particles were observed viaconfocal microscopy. Using surface initiated ATRP, Zheng and Stover114 also made P(HEMA) functionalised P(DVB) microspheres. Thus, besides improving the suspension of the P(DVB) particles in aqueous solutions, this grafting approach yields reactive particles which are capable of undergoing a variety of reactions through the hydroxyl groups. Zheng and Stover have also reported the preparation of swellable P(DVB80-co-HEMA) microspheres which they modified into ATRP initiators through the hydroxyl groups and then used them to initiate polymerization of various methacrylate monomers including HEMA.114,115 The surface functionalisation of organic surfaces via surface initiated ATRP of (HEMA) has also been reported by Bontempo et al.116 who functionalized P(St) and P(St)-P(EG) beads with P(HEMA).
Polymer-clay nanocomposites with PLLA polymer brushes on clay surfaces have been reported.117 Clay was treated with an ATRP initiator which became attached to the clay surfaces via an ion exchange process. Surface initiated ATRP of HEMA resulted in Clay-P(HEMA) composite which was subsequently used to initiate the polymerization of L-lactidevia the hydroxyl groups on the surfaces giving the Clay-brush PLLA polymer composite. TGA was used to confirm the successful modification of the clay at each step with significant weight losses being observed for the polymer-clay composites when compared to the unmodified clay. Further proof was obtained by analysis of cleaved P(HEMA) and P(HEMA)-g-PLLA brush polymer by 1H NMR. The comb polymer brushes were observed by TEM and AFM.
Leinweber et al.107 have prepared P(HEMA) functionalised fused silica particles via surface initiated ATRP for use as column packing material in the capillary electrophoresis of proteins. The materials were found to separate both basic and acidic proteins with silica particles in which the P(HEMA) was cross-linked performing better than the one with uncross-linked linear chains. A report detailing the optimization of the coating process was also published by the same group wherein modifications to the process gave packing material which resulted in good reproducibility in observed retention times for their analytes as well as an improved number of plates.118
Fukuda and co-workers have reported the synthesis of concentrated P(HEMA) brushes and investigated their interaction with proteins.119,120 In their latest publication on this work,120P(HEMA) was grafted on inner surfaces of mesoporous silicavia surface initiated ATRP and the resulting hybrid was used as column packing material to study the polymer brush-protein interactions. The elution of various proteins through the column was then monitored and it was observed that the interaction of the P(HEMA) brushes with proteins was low on outer surfaces of the material but significant on the inside. The observed interaction on the inside was attributed to distribution/fluctuation in grafting density on the material and penetration of the brushes by small proteins due to their shapes which resulted in longer elution times.
The preparation of silica nanoparticles with comb-coil polymer brushes on their surfaces was reported by Zhao et al.121 wherein the surfaces of silica nanoparticles were converted into ATRP initiators for the polymerization of HEMA followed by simultaneous ROP of L-lactide and ATRP of BA (Fig. 10). Thermogravimetric analysis confirmed the success of the grafting process and in addition the particles were also observed viaTEM wherein particle diameters of 20–30 nm were observed.
 | ||
| Fig. 10 Preparation of P(HEMA-g-LLA)-b-P(BA) coated silica particles (adapted from ref. 121). | ||
The synthesis of silica functionalised with P(HEMA) brushes has also been reported.122Bromide functionalised silica gel was used to initiate ATRP of HEMA to obtain spherical silica-P(HEMA) hybrids which were observed viaSEM. The grafting was confirmed by XPS, FT-IR and TGA.
Armes and co-workers123 reported the preparation of silica sols using anionic polyelectrolyte ATRP macroinitiators which were synthesised by the ATRP of HEMA followed by partial esterification of the P(HEMA) with 2-bromoisobutyryl bromide then complete esterification with SBA. The macroinitiators were then electrostatically adsorbed onto positively charged silica at neutral pH to obtain sols that were used to initiate polymerization of various monomers in aqueous solution.
Neoh and co-workers124 reported the synthesis of polymer-silicon (Si(100)/Si(111)) hybrids through surface initiated ATRP in which chloride/bromide functionalized silica was used to initiate polymerization of HEMA. This approach was later adapted to the preparation of polymer-silicon hybrids via interface initiated RAFT polymerization.125 In this work, the silica surface was functionalized with chlorides using vinyl benzyl chloride followed by the modification of the chloride functionalities into RAFT CTAs which served to control the polymerization of HEMA. Surface modification was confirmed by X-ray photoelectron spectroscopy, time of flight secondary ion mass spectroscopy (ToF-SIMS) and Ar beam etching. In both reports, the thickness of polymer layer was observed to depend on polymerization time.
Neoh and co-workers126 also reported the micropatterning of polymer brushes on Si(100) using a combination of surface initiated ATRP and RAFT polymerization. ATRP initiation sites were micropatterned on selected areas of a Si(100) surface by UV-induced hydrosilylation of VBC in the presence of a photomask with undesired areas covered. The polymerization of sodium styrene sulfonate (NaStS) was then undertaken to obtain Si(100) micropatterned with P(NaStS) brushes. The polymer free areas were then modified into an azo initiators followed by the RAFT polymerization of HEMA to afford Si(100) micropatterned with P(NaStS) and P(HEMA) brushes (Fig. 11). Micropatterning and grafting was ascertained by XPS and AFM.
 | ||
| Fig. 11 Non-lithographic micropatterning using ATRP and RAFT polymerization (adapted from ref. 126). | ||
Zhang and Ortiz127 reported the synthesis of P(HEMA)-g-P(EG) copolymers that were grafted on gold coated silicon. Thiol terminated P(HEMA)-g-P(EG) copolymers were first synthesised by undertaking ATRP of HEMA and P(EG) methyl ether methacrylate using a 2-(2,4-dinitrophenylthio)ethyl 2-bromo-2-methylpropionate as an initiator followed by end group modification of the polymers with mercaptoethanol. The thiol terminated polymers were then attached onto the gold coated surfaces to achieve low grafting densities resulting in mushroom like structures which were observed by AFM.
Rakhmatullina et al.128 have reported the preparation of polymeric membranes grafted from gold surfaces using surface initiated ATRP. Triblock copolymer brushes of P(HEMA)-b-P(BMA)-b-P(HEMA) were grown from bromine functionalised gold surfaces forming a polymeric membrane layer which was solvent responsive due to the amphiphilic nature of the copolymer. Immersing the gold-polymer hybrid in ethanol gave relatively thick films due to stretching of polymer chains in good solvent whilst very thin films were observed in hexane as the polymer chains collapsed since hexane is a bad solvent for all the blocks. Brush-like structures were observed when the hybrid was placed in water/ethanol mixture. The grafting process was monitored by FT-IR, contact angle measurements and 1H NMR after cleavage of the polymer from the gold surfaces.
Gold surfaces covered with partially fluorinated P(HEMA) polymers have were reported by Bantz et al.129 In this work, P(HEMA) was grown from Br functionalised gold surfaces via surface initiated ATRP followed by acylation of the P(HEMA) brushes with perfluoroalkyl acid chlorides. The contact time for acylation was carefully controlled in order to obtain various degrees of acylation, which ranged from 0 to 0.8. The success of the grafting process and acylation were then confirmed by FT-IR before the effects of fluorination on various properties were investigated. It was observed that fluorination had an effect on electrochemical barrier properties and surface wettability.
Chen et al.130 reported the preparation of hybrid hollow structures using Pickering emulsion interface initiated ATRP. In this work, ATRP initiators were immobilised onto surfaces of silica nanoparticles followed by the preparation of Pickering emulsions with the nanoparticles, oil and water. Polymerization of HEMA was then carried out by ATRP with the initiation taking place from the part of the silica surfaces exposed to water. The polymerization of HEMA led to the formation of lightly cross-linked P(HEMA) brushes and thus hybrid hollow capsules were formed. The grafting was confirmed by TGA and the hybrid hollow capsules were observed by confocal laser microscopy and optical microscopy. These particles were observed to be semi-permeable based on diffusion studies conducted with a azo dye in different solvents.
The grafting of P(HEMA) based block copolymers onto fibers using ATRP has been reported. Hou and Zhou131 reported the grafting of P(St)-b-P(HEMA) copolymers onto carbon fibers to obtain materials with improved applicability. Herein, the surface of carbon fibers was modified into ATRP initiatorsviaoxidation and acylation with thionyl chloride followed by polymerization of styrene then HEMA. The grafting was confirmed by FT-IR,TGA and SEM studies. Calmark and Malstrom132 modified the surface of cellulose into ATRP initiators followed by polymerization of MA then HEMA to obtain cellulose-g-P(MA)-b-P(HEMA) hybrids. The surface modification was confirmed by AFM and FT-IR.
Polymer metal hybrids derived from P(St)-b-P(HEMA) were reported by Wang et al.133 The block copolymers were synthesised by ATRP and were observed to form spherical core-shell micelle structures upon evaporation of dissolving solvent. To obtain the hybrids, formation of the aggregates was carried out in the presence of cobalt or nickel ions which were found to affect the morphology. The observed morphologies of the polymer-metal aggregates were related to their binding constants with oxygen in the HEMA block.
Silica-polymer-metal hybrids were also reported by Xu et al.134 They synthesised P(HEMA) and P(acrylamide)-b-P(HEMA) polymersvia surface initiated ATRP from silica surfaces and the resulting products were dispersed in Cd(II) and Cu(II) solutions to form silica-polymer-metal hybrids. Thermal analysis of the silica-polymer-metal hybrids showed that they had better stability than the silica-polymer hybrids suggesting that the introduction of the metal into improved their stability. In addition, the metal hybrids were observed to have paramagnetism by electron spin resonance studies.
The preparation of polymer brushes containing metal nanoparticles has been reported.135 Herein, a silicon surface was modified into an ATRP initiator from which P(HEMA) brushes were grown viaATRP of HEMA followed by the addition of Cu(II) ions to form polymer-metal complexes. Reduction of the complexes with sodium borohydride resulted in the formation of polymer brushes containing spherical metal nanoparticles of Cu0 which was confirmed by AFM and XPS studies.
P(HEMA)-peptide (GRGDS) hybrids with cell adhesion properties have been reported.137 The hybrids were synthesised via solid supported peptide and polymer syntheses in which the peptide was synthesised via solid phase peptide synthesis (SPPS) and modified into an ATRP initiator whilst on the solid support followed by the polymerization of HEMA. Cleavage from the solid support gave the desired product which was observed to have cell adhesion properties which are non-existent in pure P(HEMA) (Fig. 12). Neoh and co-workers136 have also reported the improvement of cell adhesion of P(HEMA) based materials by grafting collagen onto P(HEMA) grown from silicon surfaces. They demonstrated that silicon-g-P(HEMA) exhibited poor cell adhesion and growth whilst silicon-g-P(HEMA)-g-collagen had the opposite behaviour. The extent of cell adhesion was found to be dependant on the amount of grafted collagen and these materials can potentially be used in silicon-based implantable devices. Tsukagoshi et al.138 have also reported the surface functionalisation of silica with P(HEMA) and undertook protein (albumin and fibrinogen) adsorption studies.
 | ||
| Fig. 12 Synthesis of GRGDS-b-P(HEMA) hybrids via solid phase peptide synthesis (SPPS) (adapted from ref. 136). | ||
The surface functionalisation of titanium particles to obtain titanium with antibacterial and good cell adhesion properties was reported by Neoh and co-workers.139P(HEMA) was grown on titanium surfaces via surface initiated ATRP followed by modification of the polymerhydroxyl groups into amines or carboxylic acids to allow for subsequent functionalisation with penicillin, gentamicin or collagen. The bioactivity and cell adhesion of the hybrid materials were determined via a fibroplast cell culture study. It was observed that titanium-g-P(HEMA)-g-penicillin and titanium-g-P(HEMA)-g-gentamicin had antibacterial properties prohibiting cell adhesion and growth whilst titanium-g-P(HEMA)-g-collagen allowed good adhesion and cell growth. The adhesion and cell growth properties of titanium-g-P(HEMA)-g-collagen were further supported by cell culture studies of the material with osteoblast. Thus the process is useful for modifying titanium for biomedical applications.
Mei et al.140 reported the preparation of gradient P(HEMA) grafted surfaces for variable cell adhesion. They developed a method to create low density initiator gradient on silicon wafer from which the polymerization of HEMA by ATRP was initiated to obtain gradient surfaces. The gradient nature of the surface was assessed by cell adhesion studies which showed more cell adhesion on the ends with fewer polymer chains and less cell adhesion on the densely grafted ends.
Glass-polymer hybrids with bacteria recognition capabilities were reported by Chehimi and co-workers.141 Herein, glass surface was modified with bromine groups followed by surface initiated ATRP of HEMA. Success of the grafting process was confirmed by XPS and also contact angle measurements which revealed the surface was hydrophilic as evidenced from the small contact angle values. Incubation of the glass-polymer hydrid with Salmonella showed no bacteria adhered to the surface indicating its antifouling properties. However, when anti-Salmonella antibodies where attached to the hybrid viatrichlorotriazine coupling, incubation with Salmonella resulted in attachment of the bacteria on the surfaces via biorecognition of the bacteria by antibodies.
Lately, Wong and Krull142 have reported the preparation of mixed P(HEMA)/oligonucleotide films on glass and silicon surfaces. The mixing was undertaken in order to regulate the selectivity of the oligonucleotide biosensor towards detection of multiple base pair mismatches. First the surfaces were partially modified with maleimide containing molecules which allowed for the immobilisation of thiol functionalised oligonucleotidesviathiol-maeimide coupling reactions. The remaining amine groups on the surfaces were then converted into ATRP initiators followed by surface initiated ATRP of HEMA to obtain a mixed P(HEMA)/oligonucleotide brush layer. The success of the reactions was confirmed by XPS, AFM and ToF-SIMS. Their results suggested there was an improvement in the biosensing selectivity which they attributed to reduced oligonucleotide-oligonucleotide interactions due the presence of P(HEMA) chains in the brush layers.
Temperature and pH-responsive block copolymer brushes were reported by Guo and co-workers.143 The P(HEMA)-b-P(NiPAAM) copolymer was synthesized by surface initiated ATRP from a silicon substrate followed by esterification of the P(HEMA) block with succinic anhydride to obtain pH-responsive P(SEMA). Stimuli responsiveness of P(HEMA)-b-P(SEMA) was confirmed by AFM studies wherein relatively large film thicknesses were observed to at high pH due to the P(SEMA) block which was negatively charged and stretched to minimize charge repulsion. The P(NiPAAM) was established to form the top layer at low pH values regardless of the temperature whilst at high pH P(NiPAAM) only formed the top layer at low temperature with P(SEMA) being the top layer at high temperature.
Water soluble temperature responsive polymer-peptide nucleic acid (PNA) hybrids have been reported in which PNA was modified into ATRP initiators and used to polymerize HEA.144 In the presence of an aromatic molecule with three PNA arms, the PNA-P(HEA) hybrids reversibly formed 3-arm star shaped hydrid polymersvia complementary base pairing which could be turned on or off by changing the temperature (Fig. 13). The system was reported to be potentially suited for production of thermally reversible gels. Water soluble polymer-protein hybrids based on P(HEA) made viaRAFT polymerization have also been reported.145
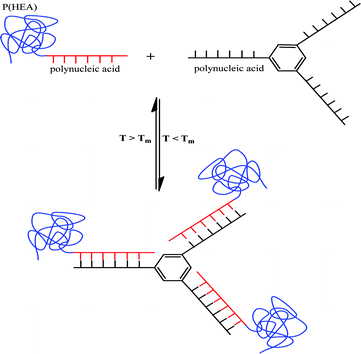 | ||
| Fig. 13 Formation of three-arm star like hybrids via reversible nucleic acid base pairing (adapted from ref. 144). | ||
Recently, the synthesis of surface attached DNA polymer bioconjugates by RAFT polymerization was reported by the He research group.146 In their work, DNA-NH2 was modified into a RAFT CTA by reacting a succinimide functionalized CTA with the amine functionalized DNA followed by the attachment of the DNA-CTA onto gold surfaces viathiol groups on DNA. The polymerization of HEMA was then undertaken to obtain Au-DNA-P(HEMA) conjugates and the success of the reaction was determined by FT-IR as well as ellipsometric characterisation. This research group has also reported the preparation Au-DNA-P(HEMA) conjugates by ATRP in which accelerated polymerization reactions were observed due to the presence of DNA.147Au-DNA-P(HEMA) based hybrids made by ATRP in electrochemical biosensing148 and DNA point mutation detection149 systems have also be reported by the same group.
Maynard and co-workers150 have reported the preparation of polymer-protein conjugates viaATRP wherein a pyridyl disulfide (PDS) functionalised ATRP initiator was used to initiate HEMA polymerization followed by attachment of bovine serum albumin (BSA) through the reduction of PDS. Good control was achieved for the polymerizations with PDIs of 1.2 being achieved. The formation of conjugates was confirmed by Ellman's test and gel electrophoresis studies. A few years later, the same group went on to introduce a method for the synthesis of polymer-peptide conjugates starting from amino acid ATRP initiators.151Polymerizations of HEMA from Fmoc-O-(2-bromoisobutyryl)-serine tert-butyl ester gave conjugates with low PDIs indicating the effectiveness of this method in producing biohybrids.
Our group has shown that self assembling cyclic peptide polymer conjugates can be prepared by ‘clicking’ alkyne-terminated PHEA prepared by RAFT polymerisation to an azide functionalised D-alt-L cyclic octapeptide via the Huisgen 1,3-dipolar cycloaddition reaction.152 Due to the high graft density, the efficiency of the click chemistry conjugation reaction was found to be highly dependent on the size of the polymer. At relatively low molecular weights as many as four polymer chains could be grafted to each cyclic peptide ring and the resulting conjugates were observed to self assemble into peptide-polymer nanotubes.
Network gels (nano/microgels)
Gel networks are cross-linked polymeric networks filled with solvent and have sizes ranging from a few nanometers to several micrometers. These materials have attracted research interest because of their applications in areas such as drug delivery, cell/enzyme immobilization, sensor design, template synthesis of nanoparticles, microreactors, artificial muscles, coatings and separation media.153–156Thermally responsive microgels have been reported by Hu et al.157 who prepared a P(NiPAAM/HEA) microgel viadispersion polymerization which was heated above the LCST of P(NiPAAM) and freeze-dried to produce a spherical microgel with P(HEA) on the surface. The surface of the microgel was then modified into a RAFT CTA and polymerization of NiPAAM was undertaken to produce thermally responsive microgel nanostructures.
Temperature and pH-responsive network hydrogels from triblock copolymers of P(DMAEMA-co-HEMA)-b-(PNiPAAM)-b-P(DMAEMA-co-HEMA) made by ATRP and cross-linked using glutaraldehyde have been reported by Neoh and co-workers.158 The cross-linking was achieved via the reaction of hydroxyl groups of P(HEMA) with glutaraldehyde. These materials were reported to have potential applications in tissue engineering and drug delivery.
Liu and co-workers159 have reported the preparation of gel networks from P(HEMA)/P(NiPAAM) triblock copolymers made by ATRP. The triblock copolymers were made using a bifunctional ATRP initiator (diethyl-meso-2,5-dibromoadipate) to construct the middle block first followed by polymerization of the second monomer. The thermoresponsive nature of the polymers was confirmed from turbidity measurements wherein the copolymers were observed to possess LCST values ranging from 21 °C–27 °C. At low temperatures, the polymers formed flower-like and branched micelles with P(HEMA) cores which aggregated to form gel networks upon heating. Inspired by the observations from their early work47 and this work, the same group has recently reported the preparation of thermoresponsive P(HEMA)-b-P(NiPAAM)-b-P(HEMA) triblock copolymers by ATRP for application in the embolisation of abnormal blood vessels.160
The preparation of gel forming cationic temperature responsive P(NiPAAM) graft copolymers was reported by Saunders and co-workers.161 Initially, diblock copolymers of DMAEMA and HEMA were made by ATRP then modified into ATRP macroinitiators by reacting the P(HEMA) hydroxyl groups with 2-bromoisobutyryl bromide. Ionic P(DMAEMA)-b-[P(HEMA)]-g-P(NiPAAM)] graft copolymers were then obtained by quaternising the P(DMAEMA) blocks of the macroinitiator copolymer with iodomethane followed by ATRP of NiPAAM. These copolymers were observed to form spherical or flower like aggregates above their LCSTs, and at high concentrations, the aggregates underwent interparticle cross-linking forming gel networks (Fig. 14).
 | ||
| Fig. 14 Synthesis of cationic graft copolymers and their self assembly at high concentration and temperature (adapted from ref. 161). | ||
Biodegradable hydrogels based on P(HEMA) which can be applied as tissue engineering scaffolds have been reported.162 P(CL) diols (α,ω-functionalised) of various molecular weights were converted into dimethacrylate macromonomers by reaction with methacryloyl chloride or into bifunctional ATRP macroinitiators by reaction with α-bromoisobutyryl bromide. The macroinitiators and macromonomers were then used to synthesise cross-linked bioresorbable P(HEMA)-co-P(CL) hydrogels by ATRP. Degradation of these hydrogels was assessed by treating the materials with either 0.007 M sodium hydroxide or phosphate buffered saline or an enzyme (lipase) and the materials were observed to degrade in sodium hydroxide as well as in presence of lipase. The degradation was observed to be dependent on the degree of cross-linking as well as molecular weight of polymers and cytotoxity studies showed the products as well as their degradation products were not cytotoxic.
Kim and co-workers163 reported the synthesis of cross-linked polymer electrolyte membranesviaATRP and esterification reactions. Two types of triblock copolymers, P(St)-b-P(HEA)-b-poly(styrene sulfonic acid) and P(St)-b-P(HEA)-b-poly(sulfopropyl methacrylate), were prepared viaATRP followed by cross-linking of the P(HEA) blocks using sulfosuccinic acid (dicarboxylic acid). The water uptake and electrical conductivity of the resulting cross-linked membranes was then investigated. The same group has also reported the preparation of proton conducting membranes which were made from comb-like copolymers.164 Poly(vinylidine fluoride-co-chlorotrifluoroethylene)-g-P(HEA) was made by ATRP and the desired membranes were obtained by cross-linking through an esterification reaction between the hydroxyl groups of P(HEA) chains with 4,5-imidazole dicarboxylic acid in the presence of phosphoric acid. The membranes were observed to exhibit proton conductivity.
Conclusions
This review has focused on the synthesis of hydroxylated (co)polymers of (P(HEMA)/P(HEA)) by LRP and applications of the resulting (co)polymers in fabrication of micelles/nanoparticles, complex polymers, hybrids and gel networks. The diversity in the terms of macromolecular architectures and nanostructured materials that can be obtained from these polymers indicates the versatility of both monomers in terms of polymerization techniques that can be used and materials that can be produced.Notes and references
- J. M. J. Fréchet, Science, 1994, 263, 1710–1715 CrossRef CAS.
- G. Ordian, Principles of Polymerization, John Wiley & Sons Inc., Singapore, 2004 Search PubMed.
- V. Coessens, T. Pintauer and K. Matyjaszewski, Prog. Polym. Sci., 2001, 26, 337–377 CrossRef CAS.
- J. V. M. Weaver, I. Bannister, K. L. Robinson, X. Bories-Azeau, S. P. Armes, M. Smallridge and P. McKenna, Macromolecules, 2004, 37, 2395–2403 CrossRef CAS.
- O. Wichterle and D. Lim, Nature, 1960, 185, 117–118 CrossRef.
- J.-P. Montheard, M. Chatzopoulos and D. Chappard, Polym. Rev., 1992, 32, 1–34 Search PubMed.
- H. F. Mark, ed., Encyclopedia of polymer science and technology, Wiley, New Jersey, 2007 Search PubMed.
- T. Jun, C. Li, F. Yuan, W. Caifeng and C. Su, J. Polym. Sci., Part A: Polym. Chem., 48, pp. 823–831 Search PubMed.
- A. Bengi, S. Mehmet, inodot, V. Elif, D. S. Nurdan and U. Ali, J. Appl. Polym. Sci., 116, pp. 628–635 Search PubMed.
- F. M. Veronese, E. Boccu, P. Caliceti, S. Lora, M. Carenza and G. Palma, J. Bioact. Compat. Polym., 1989, 4, 42–50 CrossRef CAS.
- R. Savic, A. Eisenberg and D. Maysinger, J. Drug Targeting, 2006, 14, 343–355 Search PubMed.
- C. Barner-Kowollik, ed., Handbook of RAFT polymerization, Wiley-VCH, Weinheim, 2008 Search PubMed.
- A. Carlmark, C. Hawker, A. Hult and M. Malkoch, Chem. Soc. Rev., 2009, 38, 352–362 RSC.
- D. Lowik, L. Ayres, J. M. Smeenk and J. C. M. Van Hest, Adv. Polym. Sci., 2006, 202, 19–52.
- U. Schubert, Macromol. Symp., 2008, 267, 1–8 CrossRef CAS.
- D. W. P. M. Loewik, L. Ayres, J. M. Smeenk and H. J. C. M. Van, Adv. Polym. Sci., 2006, 202, 19–52.
- E. Vargün and A. Usanmaz, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 3957–3965 CrossRef CAS.
- K. Bian and M. F. Cunningham, Macromolecules, 2005, 38, 695–701 CrossRef CAS.
- K. L. Beers, S. Boo, S. G. Gaynor and K. Matyjaszewski, Macromolecules, 1999, 32, 5772–5776 CrossRef CAS.
- L. Zhang, K. Katapodi, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 2177–2194 CrossRef CAS.
- T. Tokuda, M. Mori and T. Yamada, J. Chromatogr., A, 1996, 722, 123–127 CrossRef CAS.
- S. Mori, Anal. Chem., 1983, 55, 2414–2416 CrossRef CAS.
- H. J. A. Philipsen, H. A. Claessens, M. Bosman, B. Klumperman and A. L. German, Chromatographia, 1998, 48, 623–630 CAS.
- M. Szwarc, Nature, 1956, 178, 1168–1169 CrossRef CAS.
- M. Szwarc, M. Levy and R. Milkovich, J. Am. Chem. Soc., 1956, 78, 2656–2657 CrossRef CAS.
- T. Otsu, M. Yoshida and T. Tazaki, Makromol. Chem. Rapid Commun., 1982, 3, 133–140 CrossRef CAS.
- M. Kato, M. Kamigaito, M. Sawamoto and T. Higashimura, Macromolecules, 1995, 28, 1721–1723 CrossRef CAS.
- J.-S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614–5615 CrossRef CAS.
- S. Coca, C. B. Jasieczek, K. L. Beers and K. Matyjaszewski, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 1417–1424 CrossRef CAS.
- K. L. Robinson, M. A. Khan, M. V. de Paz Banez, X. S. Wang and S. P. Armes, Macromolecules, 2001, 34, 3155–3158 CrossRef CAS.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661–3688 CrossRef CAS.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- P. Corpart, D. Charmot, T. Biadatti, S. Z. Zard and D. Michelet, WO 9858974, 1998.
- S. Perrier and P. Takolpuckdee, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5347–5393 CrossRef CAS.
- P. Takolpuckdee, J. Westwood, D. M. Lewis and S. Perrier, Macromol. Symp., 2004, 216, 23–35 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 379–410 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2006, 59, 669–692 CrossRef CAS.
- E. Rizzardo, M. Chen, B. Chong, G. Moad, M. Skidmore and S. H. Thang, Macromol. Symp., 2007, 248, 104–116 CrossRef CAS.
- J. T. Lai, D. Filla and R. Shea, Macromolecules, 2002, 35, 6754–6756 CrossRef CAS.
- M. H. Stenzel, Chem. Commun., 2008, 3486–3503 RSC.
- G. Riess, Prog. Polym. Sci., 2003, 28, 1107–1170 CrossRef CAS.
- E. S. Read and S. P. Armes, Chem. Commun., 2007, 3021–3035 RSC.
- Y. Chan, T. Wong, F. Byrne, M. Kavallaris and V. Bulmus, Biomacromolecules, 2008, 9, 1826–1836 CrossRef CAS.
- S. Liu, J. V. M. Weaver, M. Save and S. P. Armes, Langmuir, 2002, 18, 8350–8357 CrossRef CAS.
- Z. Ping, L. Qunfeng, Q. Aixiang, S. Jianbo and L. Mangeng, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 3312–3320 CrossRef CAS.
- L. Yong-Yong, C. Han, Z. Jing-Ling, Y. Lin, D. Yu, C. Si-Xue, Z. Xian-Zheng and Z. Ren-Xi, Adv. Mater., 2009, 21, 2402–2406 CrossRef.
- C. Zhiqiang, L. Wenguang, Y. Guixiang, Z. Xiaoli, L. Xiaoze, G. Peng and Y. Kangde, Macromol. Chem. Phys., 2006, 207, 2329–2335 CrossRef.
- S. J. Holder, N. A. A. Rossi, C. T. Yeoh, G. G. Durand, M. J. Boerakker and N. Sommerdijk, J. Mater. Chem., 2003, 13, 2771–2778 RSC.
- B. Grignard, C. Jerome, C. Calberg, C. Detrembleur and R. Jerome, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 1499–1506 CrossRef CAS.
- K. Hongliang, L. Wenyong, L. Ruigang and H. Yong, Macromol. Chem. Phys., 2008, 209, 424–430 CrossRef.
- V. Schmidt, C. Giacomelli, A. R. Brisson and R. Borsali, Mater. Sci. Eng., C, 2008, 28, 479–488 CrossRef CAS.
- H. Kakwere and S. Perrier, J. Am. Chem. Soc., 2009, 131, 1889–1895 CrossRef CAS.
- C. Barner-Kowollik, T. P. Davis and M. H. Stenzel, Aust. J. Chem., 2006, 59, 719–727 CrossRef CAS.
- W. Li and K. Matyjaszewski, J. Am. Chem. Soc., 2009, 131, 10378–10379 CrossRef CAS.
- G. H. Zheng and C. Y. Pan, Polymer, 2005, 46, 2802–2810 CrossRef CAS.
- H.-i. Lee, W. Jakubowski, K. Matyjaszewski, S. Yu and S. S. Sheiko, Macromolecules, 2006, 39, 4983–4989 CrossRef CAS.
- J. Wojciech and M. Krzysztof, Macromol. Symp., 2006, 240, 213–223 CrossRef CAS.
- W. Jakubowski and K. Matyjaszewski, Macromol. Symp., 2006, 240, 213–223 CrossRef CAS.
- S. Villarroya, J. Zhou, K. J. Thurecht and S. M. Howdle, Macromolecules, 2006, 39, 9080–9086 CrossRef CAS.
- S. Villarroya, K. Dudek, J. X. Zhou, D. J. Irvine and S. M. Howdle, J. Mater. Chem., 2008, 18, 989–997 RSC.
- X. Xuewei and H. Junlian, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5523–5529 CrossRef CAS.
- X. Xuewei and H. Junlian, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 467–476 CrossRef CAS.
- X. Xuewei, J. Zhongfan, S. Ruiming and H. Junlian, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4396–4408 CrossRef CAS.
- D. Wu, Y. Yang, X. Cheng, L. Liu, J. Tian and H. Zhao, Macromolecules, 2006, 39, 7513–7519 CrossRef CAS.
- D. X. Wu, C. Z. Zhao, J. Tian and H. Y. Zhao, Polym. Int., 2009, 58, 1335–1340 CrossRef CAS.
- Z. Chuanzhuang, W. Dongxia, H. Nan and Z. Hanying, J. Polym. Sci., Part B: Polym. Phys., 2008, 46, 589–598 CrossRef.
- D. Mecerreyes, G. Moineau, P. Dubois, R. Jérôme, J. L. Hedrick, C. J. Hawker, E. E. Malmström and M. Trollsas, Angew. Chem., Int. Ed., 1998, 37, 1274–1276 CrossRef.
- D. Mecerreyes, M. Trollsas and J. L. Hedrick, Macromolecules, 1999, 32, 8753–8759 CrossRef.
- G. Zheng and H. D. H. Stover, Macromolecules, 2003, 36, 7439–7445 CrossRef CAS.
- I. Ydens, P. Degée, P. Dubois, J. Libiszowski, A. Duda and S. Penczek, Macromol. Chem. Phys., 2003, 204, 171–179 CrossRef CAS.
- J. Zhifeng, Z. Yongfeng and Y. Deyue, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 6534–6544 CrossRef CAS.
- M. Le Hellaye, C. Lefay, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3058–3067 CrossRef CAS.
- H. Kakwere and S. Perrier, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 6396–6408 CrossRef.
- F. Chun, S. Zhong, L. Yongjun, G. Lina, Z. Yaqin, L. Guolin and H. Xiaoyu, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 1811–1824 CrossRef.
- F. Chun, S. Zhong, G. Lina, Z. Sen, L. Litao, L. Guolin and H. Xiaoyu, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5638–5651 CrossRef.
- F. Chun, L. Yongjun, Y. Dong, L. Yaogong, H. Jianhua, Z. Sujuan, L. Guolin and H. Xiaoyu, J. Polym. Sci., Part A: Polym. Chem., 48, pp. 15–23 Search PubMed.
- F. Chun, S. Zhong, Y. Dong, L. Yaogong, H. Jianhua, L. Guolin and H. Xiaoyu, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4346–4357 CrossRef.
- P. Dimitrov, P. Iyer, R. Bharadwaj, P. Mallya and T. E. Hogen-Esch, Macromolecules, 2009, 42, 6873–6877 CrossRef CAS.
- W. Ling-Shu, L. Hao, D. Yao, F. Lei, L. Jing and X. Zhi-Kang, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 92–102 CrossRef CAS.
- K. L. Beers, S. G. Gaynor, K. Matyjaszewski, S. S. Sheiko and M. Moller, Macromolecules, 1998, 31, 9413–9415 CrossRef CAS.
- S. C. Hong, D. Neugebauer, Y. Inoue, J.-F. Lutz and K. Matyjaszewski, Macromolecules, 2003, 36, 27–35 CrossRef CAS.
- G. Cheng, A. Boker, M. Zhang, G. Krausch and A. H. E. Muller, Macromolecules, 2001, 34, 6883–6888 CrossRef CAS.
- H. Gao and K. Matyjaszewski, J. Am. Chem. Soc., 2007, 129, 6633–6639 CrossRef CAS.
- K. Nawel, G. Nikhil, F. Karl, G. Giorgos, H. Nikos and S. Manfred, Macromol. Rapid Commun., 2005, 26, 1693–1697 CrossRef.
- Y. Cai, Y. Tang and S. P. Armes, Macromolecules, 2004, 37, 9728–9737 CrossRef CAS.
- X. Bories-Azeau, T. Merian, J. V. M. Weaver, S. P. Armes and H. J. W. van den Haak, Macromolecules, 2004, 37, 8903–8910 CrossRef CAS.
- X. Bories-Azeau, S. P. Armes and H. J. W. van den Haak, Macromolecules, 2004, 37, 2348–2352 CrossRef CAS.
- Y. Cai and S. P. Armes, Macromolecules, 2004, 37, 7116–7122 CrossRef CAS.
- Y. Cai and S. P. Armes, Macromolecules, 2005, 38, 271–279 CrossRef CAS.
- C.-D. Vo, P. D. Iddon and S. P. Armes, Polymer, 2007, 48, 1193–1202 CrossRef CAS.
- J. Zhang, M. R. Dubay, C. J. Houtman and S. J. Severtson, Macromolecules, 2009, 42, 5080–5090 CrossRef CAS.
- B. Rixens, R. Severac, B. Boutevin and P. Lacroix-Desmazes, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 13–24 CrossRef CAS.
- S.-i. Yusa, M. Sugahara, T. Endo and Y. Morishima, Langmuir, 2009, 25, 5258–5265 CrossRef CAS.
- J. S. Kim, H. J. Jeon, M. S. Park, Y. C. You and J. H. Youk, Eur. Polym. J., 2009, 45, 1918–1923 CrossRef CAS.
- C. Chang and C. Pugh, Macromolecules, 2001, 34, 2027–2039 CrossRef CAS.
- Y. Y. Durmaz, V. Kumbaraci, A. L. Demirel, N. Talinli and Y. Yagci, Macromolecules, 2009, 42, 3743–3749 CrossRef CAS.
- R. Saito, Y. Iijima and K. Yokoi, Macromolecules, 2006, 39, 6838–6844 CrossRef CAS.
- S. Reiko and I. Yuki, Polym. Adv. Technol., 2009, 20, 280–284 CrossRef.
- S. Reiko, Y. Kazuhiro and I. Yuki, Macromol. Symp., 2007, 249–250, 398–405.
- X. Jiang, M. C. Lok and W. E. Hennink, Bioconjugate Chem., 2007, 18, 2077–2084 CrossRef CAS.
- J. Yang, P. Zhang, L. Tang, P. Sun, W. Liu, P. Sun, A. Zuo and D. Liang, Biomaterials, 2010, 31, 144–155 CrossRef CAS.
- G. Pasut and F. M. Veronese, Prog. Polym. Sci., 2007, 32, 933–961 CrossRef CAS.
- M. J. Vicent, L. DieudonnÃ, R. J. Carbajo and A. Pineda-Lucena, Expert Opin. Drug Delivery, 2008, 5, 593–614 Search PubMed.
- R. Satchi-Fainaro, R. Duncan and C. M. Barnes, in Polymer Therapeutics II: Polymers as Drugs, Conjugates and Gene Delivery Systems, Springer-Verlag, Berlin, 2006, pp. 1–65 Search PubMed.
- J.-F. Lutz and H. G. Börner, Prog. Polym. Sci., 2008, 33, 1–39 CrossRef CAS.
- H.-A. Klok, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 1–17 CrossRef CAS.
- F. C. Leinweber, J. Stein and M. Otto, Fresenius J. Anal. Chem., 2001, 370, 781–788 CrossRef CAS.
- X. Pei, J. Hao and W. Liu, J. Phys. Chem. C, 2007, 111, 2947–2952 CrossRef CAS.
- P. Liu, Y. Liu and Z. Su, Ind. Eng. Chem. Res., 2006, 45, 2255–2260 CrossRef CAS.
- C. Wan, M. Li, X. Bai and Y. Zhang, J. Phys. Chem. C, 2009, 113, 16238–16246 CrossRef CAS.
- H. Kong, C. Gao and D. Yan, J. Am. Chem. Soc., 2004, 126, 412–413 CrossRef CAS.
- Y. Gao, X. P. Gao, Y. F. Zhou and D. Y. Yan, Nanotechnology, 2008, 19 Search PubMed.
- A. S. Goldmann, A. Walther, L. Nebhani, R. Joso, D. Ernst, K. Loos, C. Barner-Kowollik, L. Barner and A. H. E. Müller, Macromolecules, 2009, 42, 3707–3714 CrossRef CAS.
- G. Zheng and H. D. H. Stover, Macromolecules, 2003, 36, 1808–1814 CrossRef CAS.
- G. Zheng and H. D. H. Stover, Macromolecules, 2002, 35, 7612–7619 CrossRef CAS.
- D. Bontempo, N. Tirelli, G. Masci, V. Crescenzi and J. A. Hubbell, Macromol. Rapid Commun., 2002, 23, 417–422 CrossRef CAS.
- Y. Yang, D. Wu, C. Li, L. Liu, X. Cheng and H. Zhao, Polymer, 2006, 47, 7374–7381 CrossRef CAS.
- A. Feldmann, U. Claußnitzer and M. Otto, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2004, 803, 149–157 CrossRef CAS.
- C. Yoshikawa, A. Goto, Y. Tsujii, T. Fukuda, T. Kimura, K. Yamamoto and A. Kishida, Macromolecules, 2006, 39, 2284–2290 CrossRef CAS.
- Y. Chiaki, G. Atsushi, T. Yoshinobu, I. Norio, N. Kazuki and F. Takeshi, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 4795–4803 CrossRef CAS.
- H. Zhao, X. Kang and L. Liu, Macromolecules, 2005, 38, 10619–10622 CrossRef CAS.
- K. Zhang, H. Li, H. Zhang, S. Zhao, D. Wang and J. Wang, Mater. Chem. Phys., 2006, 96, 477–482 CrossRef CAS.
- C.-D. Vo, A. Schmid, S. P. Armes, K. Sakai and S. Biggs, Langmuir, 2007, 23, 408–413 CrossRef CAS.
- F. J. Xu, Q. J. Cai, E. T. Kang and K. G. Neoh, Langmuir, 2005, 21, 3221–3225 CrossRef CAS.
- Q. Peng, D. M. Y. Lai, E. T. Kang and K. G. Neoh, Macromolecules, 2006, 39, 5577–5582 CrossRef CAS.
- F. J. Xu, E. T. Kang and K. G. Neoh, J. Mater. Chem., 2006, 16, 2948–2952 RSC.
- D. Zhang and C. Ortiz, Macromolecules, 2004, 37, 4271–4282 CrossRef CAS.
- R. Ekaterina, M. Alexandre, B. Thomas, M. Violeta and M. Wolfgang, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 1–13 CrossRef CAS.
- M. R. Bantz, E. L. Brantley, R. D. Weinstein, J. Moriarty and G. K. Jennings, J. Phys. Chem. B, 2004, 108, 9787–9794 CrossRef CAS.
- C. Yunhua, W. Chaoyang, C. Jianxin, L. Xinxing and T. Zhen, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 1354–1367 CrossRef CAS.
- J. Q. Hou and X. D. Zhou, J. Macromol. Sci., Part A: Pure Appl. Chem., 2008, 45, 990–996 Search PubMed.
- A. Carlmark and E. E. Malmstrom, Biomacromolecules, 2003, 4, 1740–1745 CrossRef CAS.
- W. Yanmei, W. Jingshen, Y. Jinying, S. Guifen and P. Caiyuan, J. Appl. Polym. Sci., 2002, 83, 2883–2891 CrossRef.
- X. Yaxin, L. Hongtu, W. Di, L. Yapeng, S. Yantao, A. Peng and W. Jingyuan, J. Appl. Polym. Sci., 2007, 105, 2146–2154 CrossRef.
- K. Zhang, H. T. Li, S. Zhao, W. Wang, S. W. Wang, Y. X. Xu, W. Z. Yu and J. Y. Wang, Polym. Bull., 2006, 57, 253–259 CrossRef CAS.
- F. J. Xu, S. P. Zhong, L. Y. L. Yung, Y. W. Tong, E. T. Kang and K. G. Neoh, Tissue Eng., 2005, 11, 1736–1748 CrossRef CAS.
- Y. Mei, K. L. Beers, H. C. M. Byrd, D. L. VanderHart and N. R. Washburn, J. Am. Chem. Soc., 2004, 126, 3472–3476 CrossRef CAS.
- T. Tsukagoshi, Y. Kondo and N. Yoshino, Colloids Surf., B, 2007, 54, 101–107 CrossRef CAS.
- F. Zhang, Z. L. Shi, P. H. Chua, E. T. Kang and K. G. Neoh, Ind. Eng. Chem. Res., 2007, 46, 9077–9086 CrossRef CAS.
- Y. Mei, T. Wu, C. Xu, K. J. Langenbach, J. T. Elliott, B. D. Vogt, K. L. Beers, E. J. Amis and N. R. Washburn, Langmuir, 2005, 21, 12309–12314 CrossRef CAS.
- B. Mrabet, M. N. Nguyen, A. Majbri, S. Mahouche, M. Turmine, A. Bakhrouf and M. M. Chehimi, Surf. Sci., 2009, 603, 2422–2429 CrossRef CAS.
- A. K. Y. Wong and U. J. Krull, Anal. Chim. Acta, 2009, 639, 1–12 CrossRef CAS.
- W. Xi, X. Xing, W. Xiaohong, Z. Jianjun, L. Lin, X. Jun and G. Baohua, Macromol. Rapid Commun., 2007, 28, 828–833 CrossRef CAS.
- Y. Wang, B. A. Armitage and G. C. Berry, Macromolecules, 2005, 38, 5846–5848 CrossRef CAS.
- C. Boyer, V. Bulmus, J. Liu, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, J. Am. Chem. Soc., 2007, 129, 7145–7154 CrossRef CAS.
- P. He and L. He, Biomacromolecules, 2009, 10, 1804–1809 CrossRef CAS.
- X. Lou and L. He, Langmuir, 2006, 22, 2640–2646 CrossRef CAS.
- Y. Wu, S. Liu and L. He, Anal. Chem., 2009, 81, 7015–7021 CrossRef CAS.
- X. Lou, M. S. Lewis, C. B. Gorman and L. He, Anal. Chem., 2005, 77, 4698–4705 CrossRef CAS.
- D. Bontempo, K. L. Heredia, B. A. Fish and H. D. Maynard, J. Am. Chem. Soc., 2004, 126, 15372–15373 CrossRef CAS.
- R. M. Broyer, G. M. Quaker and H. D. Maynard, J. Am. Chem. Soc., 2008, 130, 1041–1047 CrossRef CAS.
- R. Chapman, K. A. Jolliffe and S. Perrier, Aust. J. Chem., 2010 Search PubMed , ASAP.
- M. Karg and T. Hellweg, Curr. Opin. Colloid Interface Sci., 2009, 14, 438–450 CrossRef CAS.
- A. Fernández-Barbero, I. J. Suárez, B. Sierra-Martín, A. Fernández-Nieves, F. J. de las Nieves, M. Marquez, J. Rubio-Retama and E. López-Cabarcos, Adv. Colloid Interface Sci., 2009, 147–148, 88–108 CrossRef CAS.
- M. Das, S. Mardyani, W. C. W. Chan and E. Kumacheva, Adv. Mater., 2006, 18, 80–83 CrossRef CAS.
- J. A. Trelles, F. Quiroga, C. Britos, E. E. Smolko and M. Grasselli, Radiat. Phys. Chem., 79, pp. 241–245 Search PubMed.
- T. Hu, Y. You, C. Pan and C. Wu, J. Phys. Chem. B, 2002, 106, 6659–6662 CrossRef CAS.
- F.-J. Xu, E.-T. Kang and K.-G. Neoh, Biomaterials, 2006, 27, 2787–2797 CrossRef CAS.
- Z. Xiaoli, L. Wenguang, C. Dayong, L. Xiaoze and W. L. William, Macromol. Chem. Phys., 2007, 208, 1773–1781 CrossRef CAS.
- F. Y. Dai, L. Tang, J. H. Yang, X. L. Zhao, W. G. Liu, G. Chen, F. S. Xiao and X. Q. Feng, J. Mater. Sci.: Mater. Med., 2009, 20, 967–974 CrossRef CAS.
- R. Liu, P. De Leonardis, F. Cellesi, N. Tirelli and B. R. Saunders, Langmuir, 2008, 24, 7099–7106 CrossRef CAS.
- S. Atzet, S. Curtin, P. Trinh, S. Bryant and B. Ratner, Biomacromolecules, 2008, 9, 3370–3377 CrossRef CAS.
- D. K. Lee, K. J. Lee, Y. W. Kim and J. H. Kim, Desalination, 2008, 233, 104–112 CrossRef CAS.
- Y. W. Kim, J. T. Park, J. H. Koh, D. K. Roh and J. H. Kim, J. Membr. Sci., 2008, 325, 319–325 CrossRef CAS.
Footnote |
| † This paper is part of a Polymer Chemistry issue highlighting the work of emerging investigators in the polymer chemistry field. Guest Editors: Rachel O'Reilly and Andrew Dove. |
| This journal is © The Royal Society of Chemistry 2011 |