UV wavelength-dependent DNA damage and human non-melanoma and melanoma skin cancer†
Gerd P.
Pfeifer
and
Ahmad
Besaratinia
Department of Cancer Biology, Beckman Research Institute of the City of Hope, Duarte, CA 91010, USA. E-mail: gpfeifer@coh.org; Fax: +1 626-358-7703; Tel: +1 626-301-8853
First published on 1st August 2011
Abstract
Ultraviolet (UV) irradiation from the sun has been epidemiologically and mechanistically linked to skin cancer, a spectrum of diseases of rising incidence in many human populations. Both non-melanoma and melanoma skin cancers are associated with sunlight exposure. In this review, we discuss the UV wavelength-dependent formation of the major UV-induced DNA damage products, their repair and mutagenicity and their potential involvement in sunlight-associated skin cancers. We emphasize the major role played by the cyclobutane pyrimidine dimers (CPDs) in skin cancer mutations relative to that of (6–4) photoproducts and oxidative DNA damage. Collectively, the data implicate the CPD as the DNA lesion most strongly involved in human cancers induced by sunlight.
 Dr Gerd P. Pfeifer | Dr Gerd P. Pfeifer is Professor and Chair of the Department of Cancer Biology at the Beckman Research Institute of the City of Hope in Duarte, California. For almost two decades, Dr Pfeifer has investigated the mechanisms of ultraviolet radiation induced DNA damage and mutagenesis in mammalian cells. His contributions include the development of methods for single base resolution mapping of UV damage and the demonstration that the modified DNA base 5-methylcytosine is particularly prone to form cyclobutane pyrimidine dimers. |
 Dr Ahmad Besaratinia | Dr Ahmad Besaratinia has worked with Dr Pfeifer on DNA damage and mutations induced by UVA, UVB and solar radiation. |
Introduction: solar UV radiation and skin cancer
The main types of human skin cancer are non-melanoma (basal and squamous cell carcinoma) skin cancers and melanoma skin cancers. The non-melanoma tumors originate from keratinocytes that have undergone malignant transformation. Melanoma results from transformed melanocytes in the skin. Currently, skin cancer is the most common tumor diagnosed in the United States and many other countries and the numbers of both non-melanoma and particularly those of melanoma skin cancers have increased dramatically over the last several decades.1–3 Melanomas represent less than 10% of all skin cancers, yet they account for the vast majority of skin cancer-related deaths due to the high metastatic potential and resistance to therapy of these tumors. The available epidemiological evidence indicates clearly that solar UV radiation is associated with skin cancer.4,5 Thus, skin cancers are one of the few examples of human cancer where the etiological agent is known with high certainty. These cancers thus provide a useful system to dissect the various steps of carcinogenesis, allowing the characterization of the initial events that occur at the DNA level and the identification of target genes that are frequently and specifically mutated.UV-induced DNA damage and its repair
Irradiation of purified DNA or living organisms with UV light induces the direct or indirect formation of several types of DNA lesions. The most frequent form of DNA damage induced in the UVB range (290–320 nm) or by high-energy artificial UVC (254 nm) irradiation involves the dimerization of pyrimidines. This leads to the formation of cis-syn cyclobutane pyrimidine dimers (CPDs) and pyrimidine (6–4) pyrimidone photoproducts [(6–4) photoproducts; (6–4)PPs] as the major DNA damage products (see Fig. 1). Several minor damaged DNA base products, such as certain purine dimers and pyrimidine photohydrates, are also created.6 The cyclobutane rings of CPDs are formed between the 5,6 bonds of two adjacent pyrimidine bases (thymine, cytosine, or 5-methylcytosine). (6–4)PPs arise through a complicated rearrangement and contain a stable single bond between position 6 and position 4 of two adjacent pyrimidine bases. In most experimental systems studied, the (6–4)PPs are formed at levels quite significantly lower than those of CPDs.7,8UVA (320–400 nm) irradiation may also induce CPDs in DNA9,10 albeit at rather low levels.11 In addition, through an indirect mechanism, both UVA and UVB can promote the formation of oxidized DNA bases such as 8-oxo-7,8-dihydro-2′-deoxyguanosine (8-oxo-dG) (Fig. 1).11–16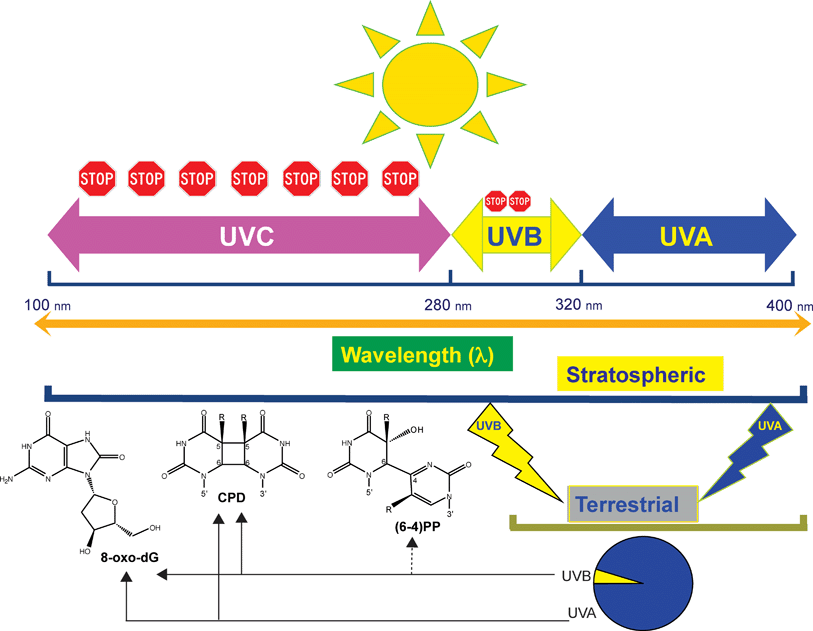 | ||
| Fig. 1 The major UVB- and UVA-induced DNA photoproducts are induced at different wavelengths. The diagram shows the subdivision of the solar UV spectrum. Shorter UV wavelengths are blocked by oxygen and ozone (stop signs). Terrestrial sunlight contains UVA and UVB components at wavelengths of ∼300 nm and longer. The specific DNA lesions produced by UVA and UVB in this range of wavelengths are indicated by arrows. | ||
There has been some disagreement regarding the relative yields of oxidative damage and cyclobutane dimers in the UVA range. Chromatographic measurements9,17,18 and analyses by the alkaline elution technique13 have shown that CPDs are more frequent than oxidized purines when large doses of UVA are used for irradiation. However, higher ratios of oxidized purines to CPDs have been reported when irradiations were carried out under defined conditions with cultured cells even in the absence of exogenous photosensitizers.11,198-Oxo-guanine is the most frequent UVA-induced oxidative base lesion, but DNA strand breaks are also generated at a lower yield, as well as lower amounts of oxidized pyrimidines.20,21 In accordance with the complexity of the lesion spectrum, the mutation spectra induced by UVA-mediated photooxidation have shown to include G to A transitions,22,23 presumably due to CPDs, as well as G to T transversions, presumably due to 8-oxodG.19,24 The basal layer in human squamous cell tumors harbored more UVA (G to T) than UVB (C to T) fingerprint mutations, which suggested a role for UVA-induced oxidized guanines in human skin carcinogenesis.25 The lesion and mutation spectra seem to vary with the experimental system used and may depend on cell type and irradiation conditions. Importantly, UVA is much less mutagenic than UVB and it is often difficult to establish an appropriate dose response for mutation induction by UVA.
Dimerized base-containing DNA lesions such as UV photoproducts are generally repaired by the nucleotide excision repair (NER) pathway. This DNA repair system plays an important role in preventing UV-induced skin cancers. Several rare human genetic disorders that include xeroderma pigmentosum (XP) and Cockayne syndrome are characterized by a defect in nucleotide excision repair. Cells from the affected patients are hypersensitive to UV light. The incidence of skin cancer of all types (including melanoma) is increased by several orders of magnitude in XP patients26,27 suggesting that pyrimidine dimers contribute to melanoma and non-melanoma skin tumors. Although repair of (6–4)PPs in normal cells is generally much faster than that of CPDs,7 repair of both types of photoproducts is absent in many of the skin cancer-prone XP patients. Thus, the dramatically increased incidence of skin cancer in nucleotide excision repair- defective patients is one piece of strong evidence that links dimeric DNA photoproducts to skin cancer. On the other hand, defects in base excision repair genes that would result in impaired repair of oxidative base lesions, have not been linked in a major way to skin cancer in human populations.
Mapping of UV damage in human genes: relation to skin cancer mutations
In our work on UV photoproducts over the past two decades, we have attempted to relate UV-induced DNA damage to the specific genetic changes that are observed in human skin cancers. Using the ligation-mediated polymerase chain reaction (LMPCR) technique developed for mapping of UV damage,28,29 we have analyzed the distribution and repair of UV-photoproducts along several human genes at the sequence level.30–33 In some of our studies, we focused on the human tumor suppressor gene p53, which contains mutations in a high percentage of human non-melanoma skin cancers.34–37 We found that sites of mutation hotspots coincide with nucleotide positions where the DNA repair process for removal of UV-induced CPDs is particularly slow.31 In addition, we showed that the sequence distribution of CPDs induced by natural sunlight is very different from that induced by UVC (254 nm) at sequence positions specifically containing 5-methylcytosine bases. The sunlight-induced CPD distribution provided a good match with skin cancer mutations.38 The results suggest that sunlight-induced formation of CPDs and their reduced DNA repair efficiency may strongly contribute to the mutation spectrum in the p53 gene of non-melanoma skin cancers. 5-methylcytosines are present almost exclusively at CpG dinucleotides in mammalian cells and all CpGs in the p53 coding sequence are highly methylated.39The increased mutability of CpG dinucleotides usually is attributed to two different mechanistic pathways: the spontaneous deamination of 5-methylcytosine and the preferential targeting of methylated CpG sequences by carcinogens.40 In skin cancers, C→T transition mutations are particularly frequent at the trinucleotide sequences 5′-PymCG (5′TmCG and 5′CmCG) in the p53 gene. These sequences contain 5-methylcytosines in human keratinocytes,39 and they are the preferential sites for CPD formation when sunlight is used for irradiation.38 Mechanistically, 5-methylcytosine has a longer wavelength absorption maximum relative to cytosine and a tenfold increase of the fluorescence lifetime, making excited state reactions more probable.41 These parameters most likely provide explanations for the preferential susceptibility of dipyrimidines containing 5-methylcytosine to sunlight- or UVB-induced CPD formation. Using mammalian mutational reporter genes, it was demonstrated that 5-methylcytosines contribute significantly to sunlight induced mutational hotspots in mammalian cells.42–44 One important mechanism for the selective mutagenesis of 5′-PymCG sites by UVB is the efficient deamination of 5-methylcytosines within CPDs, an event that can then be followed by “correct” polymerase bypass leading to a mutation.45 The available evidence clearly indicates that 5-methylcytosine plays a major role in UVB mutagenesis in mammalian cells. The finding that dipyrimidines containing 5-methylcytosine (5mC) rather than the commonly studied thymine-thymine (T–T) dimers are the most frequent lesions induced by UVB in sunlight has biological significance.38
Wavelength-dependence of UV photoproduct formation
The UV spectrum of sunlight is conventionally divided into short wavelength UVC (λ < 280 nm), mid-wavelength UVB (λ 290–320 nm), and long wavelength UVA (λ > 320–400 nm).46 The UVC fraction of sunlight is entirely absorbed by stratospheric oxygen (O2), which subsequently undergoes decomposition and recombination reactions, giving rise to ozone (O3). The resulting O3 molecules can function as a filter and absorb the majority of sunlight UVB.47,48 Thus, the solar UV wavebands that reach the surface of the earth, and as such are of relevance for photocarcinogenesis, are UVA and UVB, which comprise 95% and 5%, respectively, of the terrestrial sunlight UV48,49 (Fig. 1).To determine the contribution of specific UV wavelengths to solar mutagenesis, we used a tunable near-monochromatic laser system to investigate the induction of DNA damage, both in the overall genome and at the nucleotide resolution level, in the genomic DNA of transgenic Big Blue® mouse fibroblasts irradiated with thirteen different, yet, precisely defined serial wavelengths, inclusive of UVC, UVB, and UVA. Subsequently, we sought correlation between the locations of UV-induced DNA lesions in the cII transgene of irradiated DNA samples and the frequency distribution and codon position of the induced cII mutations in mouse cells irradiated with simulated sunlight (SSL).50 Using a combination of enzymatic digestion assays coupled with gel electrophoresis, immunodot blot assays, and DNA footprinting, we demonstrated a unique wavelength-dependent formation of both CPDs and (6–4)PPs, based on direct UV absorption of DNA, in irradiated mouse genomic DNA, which could partially explain the induction of mutations in mouse cells irradiated with SSL.
More specifically, UV absorption of DNA accounted for nearly all the induced lesions in irradiated genomic DNA; however, only one-half of all sunlight-induced cII-mutated codons in SSL-irradiated cells co-localized with the preferential sites of lesion-formation found in UV-irradiated purified genomic DNA. There are several explanations for these findings including chromatin-dependent lesion formation, site-specific repair in vivo, site-specific deamination, or polymerase bypass, as well as selection for phenotypically recoverable mutations. However, these findings may also imply that many of the sunlight-induced mutations found in SSL-irradiated mouse cells might be ascribed to lesions that are not generated through direct UV absorption of DNA in the UVA-UVB range. These lesions may have arisen from photosensitization reactions, mostly in the UVA range,51–54 which would implicate UVA as a relevant contributor to solar mutagenesis.
In addition to the wavelength-dependent formation of both CPDs and (6–4)PPs, we also found a divergence of formation of these lesions at wavelength 296 nm of irradiation in mouse genomic DNA. Whereas substantial formation of (6–4)PPs was detectable in samples irradiated at this wavelength, which intensified as the irradiation wavelength decreased, only small quantities of these lesions were found in samples irradiated at wavelengths 300–305 nm with no detectable level of (6–4)PPs in samples irradiated with longer wavelengths. Although CPD formation followed the same pattern of increase with decreasing wavelengths of irradiation, there were substantial levels of CPDs in samples irradiated with UVB wavelengths bordering the UVA range, and small but detectable levels of these lesions in samples irradiated with longer wavelengths. Whereas the unique wavelength-dependent formation of both photodimeric lesions deserves special attention, the distinct formation of CPDs as compared to (6–4)PPs at longer UV wavelengths concurs with the predominant formation of CPDs in sunlight-irradiated DNA or cells.55–57 Because the terrestrial sunlight spectrum rolls off sharply at wavelength ∼300 nm (e.g., ref. 58), the relatively low detection of (6–4)PPs at around this wavelength or the longer wavelengths in irradiated genomic DNA suggests that CPDs are the principal lesion accounting for most DNA damage-dependent biological effects of current day sunlight. There is concern, however, that the ongoing depletion of the stratospheric ozone layer may cause a shift in the DNA damage spectrum on the earth's surface.
UV mutagenesis
UV mutagenesis is characterized by a high frequency of transition mutations at dipyrimidine sequences that contain cytosine bases suggesting that dimeric pyrimidine photoproducts containing cytosine are highly mutagenic lesions.53 However, the precise mechanism of how UV light causes C to T transition mutations at dipyrimidine sites is still not completely resolved. The CPD is believed to be the major mutagenic lesion in mammalian cells owing to its high level of induction, slow repair by NER and efficient replication bypass tolerance. However, the mutation frequency obtained with a single site-specific 5′-TT-CPD is generally low.59–62 This is consistent with the infrequent recovery of mutations at 5′-TT sequences from UV-irradiated cells. Importantly, the mutagenicity of a site-specific CPD containing the 5′-TC sequence also is very low with >95% accurate lesion bypass,63 and the bypass of a CPD-containing 5-methylcytosine is even more than 99% accurate.64 The exact contribution of (6–4)PPs to UV mutagenesis is essentially unknown6,7,65 and may strongly depend on wavelength.50 Although (6–4)PPs are mutagenic when studied as single site-specific lesions in vitro or in transfection experiments, their direct contribution to solar UV mutagenesis in mammalian cells is probably minor. This is because they are repaired much more rapidly than the CPD photoproducts53,65,66 and because they are induced only at very low levels by wavelengths in terrestrial sunlight.50 Importantly, using a set of mutation reporter cell lines expressing exogenous photolyase genes [either CPD-specific photolyase or (6–4)-specific photolyase] we have shown that the CPDs are responsible for >80% of the UVB-induced mutations in DNA repair-proficient mammalian cells.43 However, the ability of (6–4)PPs to block DNA replication and transcription is much greater than that of the CPD.7,67 Therefore, these lesions may contribute to UVB-induced cell killing and may be involved in other detrimental cellular responses including induction of altered gene expression patterns and chromosomal rearrangements.The low frequency of mutations recovered at 5′-TT sequences in UV-irradiated cells is likely due to the precise action of DNA polymerase eta, which correctly bypasses these lesions in vitro and incorporates two adenines opposite the lesion. DNA polymerase eta is encoded by the RAD30 gene in yeast and by the XPVgene in humans.68–70In vivo, the UV-induced DNA lesion may be initially encountered by a replicative polymerase, which may or may not be able to efficiently insert nucleotides opposite the damaged site. If it cannot insert, the replicative polymerase will dissociate from the primer/template. This may clear the way for a specialized DNA damage bypass polymerase. Unlike the replicative polymerases, these bypass polymerases can synthesize past a variety of DNA lesions and have a very low fidelity and processivity on undamaged templates. One of them, DNA polymerase eta, is defective in XP-V cells, and strong evidence suggests that this polymerase carries out mostly error-free translesion synthesis (TLS) past CPDs in yeast and in mammalian cells.71–73
At least two models of UV mutagenesis can be proposed to explain the preponderance of C→T transition mutations in UV-irradiated cells. One pathway involves direct lesion bypass by a DNA polymerase that incorporates adenines opposite a cytosine (C) or 5-methylcytosine (mC) within the dimer. The nature of such an error-prone polymerase in vivo is not exactly clear but may include DNA polymerases zeta and kappa.73 The second pathway is represented by a model, in which the C or 5mC first deaminates within the CPD lesion. The deamination reaction is then followed by ‘correct’ lesion bypass during DNA replication by DNA polymerase eta. We and others have provided evidence that the second pathway, the deamination bypass pathway, may play an important role in UVB mutagenesis in mammalian cells.45,64,74–76
Ultraviolet radiation (UVA and UVB) and melanoma
The causative relationship between UVB (290–320 nm) and non-melanoma skin cancer is well documented.77 However, the link between sunlight exposure and melanoma is less clear and is still controversial.4,78 Melanoma, the most lethal form of human skin cancer, shows a dramatic rate of increase worldwide and causes several thousand deaths each year in the United States.79,80 There is epidemiological evidence – albeit not without controversy – that a history of sunburn and intermittent exposure to strong sunlight, particularly during childhood and young adolescence, may promote the formation of melanoma.81–83 Various epidemiological studies have addressed the relative contributions of UVB (290–320 nm) and UVA radiation (320 to 400 nm) to skin carcinogenesis, in particular to the development of melanoma.46,84–87UVB is clearly linked to non-melanoma skin cancers and UVA may play a role in melanoma although the latter association is controversial.UVB radiation induces squamous-cell carcinoma in mice.88UVA radiation is also a complete rodent carcinogen.89 Although UVB dominates the carcinogenic effects of sunlight, UVA is estimated to contribute 10–20% to the carcinogenic dose.90 The use of tanning salons has now become a popular practice, particularly in geographic areas of Northern latitude. Certain high-powered tanning lamps emit mainly long wave UVA (340–400 nm, also known as UVA1). Exposure to sunlamps or sunbeds has been linked to a 2-fold risk increase for melanoma.91 This was confirmed in a more recent meta analysis in which a risk factor of 1.75 was determined for sunbed use before 35 years of age.92UVA radiation is far more abundant in sunlight than is UVB (>20 times radiant energy). An additional concern regarding the possible carcinogenic effect of UVA is the use of sunscreens that protect effectively against erythema-inducing UVB wavelengths yet do allow significant penetration of UVA or contain unstable UVA filters, thus lengthening the time a susceptible person can spend in the sun and still is exposed to high doses of UVA. Another relevant issue is that DNA damage occurs even at suberythemal UV doses and that prevention of sunburn does not necessarily mean prevention of skin cancer.
Free radicals, in particular reactive oxygen species, can be generated by UVA irradiation and can damage DNA. UVA causes substantial levels of oxidative DNA damage that is potentially mutagenic and can induce G to T transversions.11,19 Oxidized guanines produced by UVA are repaired quickly and much more rapidly than CPDs.66 Thus, at least in slowly dividing cells, their contribution to mutagenesis is expected to be much smaller than that of CPDs. UVA also induces low levels of CPDs.9–11,18 In contrast to UVB, the low-energy UVA radiation is poorly absorbed by DNA. Hence, the absorption by other molecules (endogenous photosensitizers) becomes important24 although a direct photochemical mechanism for CPD induction by UVA has also been described.93,94UVA is capable of inducing melanoma-like lesions in marsupials95 and this had also been reported earlier for certain fish species.96 However, in more recent studies with Xiphophorus fish and also in a mouse model an effect of UVA on melanoma induction has not been observed.97,98
A few specific gene targets of sunlight in melanoma have been identified. By studying the types of mutations in these genes, some clues as to UV-induced origins of the mutations can be obtained. The p16/CDKN2 gene often carries germ-line mutations in melanoma-susceptible families.99 Sporadic (non-familial) melanomas also can carry somatic missense mutations in the CDKN2 gene.100 Although the finding of common UV-signature C→T transition mutations in the CDKN2 gene of melanomas suggested targeting of this gene by UVB irradiation,66,101 the observation of similar C→T mutations in several other tumors not related to sunlight may call this association into question. Nonetheless, the available data suggest that CDKN2 may be an important UVB target in melanoma formation. Another gene frequently mutated in melanoma is BRAF, which is mutated in up to 60–70% of melanomas. The most common mutation found in BRAF is a T to A transversion that is not at a dipyrimidine site. A potential sunlight-induced origin of this mutation has been discussed102,103 but remains unproven. The mutation may largely be driven by selection of specific mutant forms of the BRAF oncogene. On the other hand, mutations in the GNAQ and GNA11 genes found in uveal melanomas104 do carry the signature of pyrimidine dimer-induced C to T transition mutations.105 Of note, XP patients also exhibit higher frequency of melanoma, even though mutation spectra in some genes of these tumors do not always harbor a pyrimidine dimer signature. This may suggest that other unidentified bulky lesions are involved or that dimers play a role only in the initiation phase of the disease.
In addition to the specific relationship of DNA damage-induced mutagenesis and melanoma mutations, it should be kept in mind, however, that melanoma differs from other skin cancers in many physiological parameters.106 For example, melanocytes are characterized by their resistance to apoptosis107 and long-term survival in the skin and in these respects differ quite substantially from other cells such as keratinocytes.
Whole genome sequencing data will help unravel melanoma etiology
With the recent advent of high-throughput DNA sequencing, we will see much additional data that can provide clues as to the sunlight-specific origin of melanoma. Today, the data are still limited but are beginning to provide interesting insight. In one of the first studies dealing with the sequencing of an entire cancer genome, Pleasance et al. sequenced the genome of a melanoma cell line using Illumina short sequence read technology.108 More than 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 base substitutions were identified in the tumor relative to germ line DNA. By far the most common mutation observed was the C to T mutation accounting for more than two thirds of all mutations in this melanoma sample. Interestingly, a total of 92% of the C to T mutations occurred at the 3′ base of a pyrimidine dinucleotide, a number much higher than expected by chance. Such mutations are characteristic of UVB-induced DNA damage. In addition, both C to T substitutions (7.7%) and CC to TT double substitutions (10.0%) showed elevated frequencies at CpG dinucleotides compared to that expected by chance (4.4%). The mutation spectrum and sequence context suggested that most C to T somatic substitutions in this melanoma cell line can be attributed to ultraviolet-light-induced DNA damage.109 Notably, the mutational spectrum of this melanoma cell line was also shaped by DNA repair events. The specialized mechanism of transcription-coupled nucleotide excision repair removes CPDs preferentially from the transcribed strand of active genes.110 This strand-specific repair was reflected in the distribution of C to T and CC to TT mutations in the melanoma genome, in which these types of mutations were more prominent on the non-transcribed DNA strand of active genes.
000 base substitutions were identified in the tumor relative to germ line DNA. By far the most common mutation observed was the C to T mutation accounting for more than two thirds of all mutations in this melanoma sample. Interestingly, a total of 92% of the C to T mutations occurred at the 3′ base of a pyrimidine dinucleotide, a number much higher than expected by chance. Such mutations are characteristic of UVB-induced DNA damage. In addition, both C to T substitutions (7.7%) and CC to TT double substitutions (10.0%) showed elevated frequencies at CpG dinucleotides compared to that expected by chance (4.4%). The mutation spectrum and sequence context suggested that most C to T somatic substitutions in this melanoma cell line can be attributed to ultraviolet-light-induced DNA damage.109 Notably, the mutational spectrum of this melanoma cell line was also shaped by DNA repair events. The specialized mechanism of transcription-coupled nucleotide excision repair removes CPDs preferentially from the transcribed strand of active genes.110 This strand-specific repair was reflected in the distribution of C to T and CC to TT mutations in the melanoma genome, in which these types of mutations were more prominent on the non-transcribed DNA strand of active genes.
Sequencing of 518 protein kinase genes in six melanoma samples uncovered 144 mutations and more than 90% of these mutations were C to T transitions at dipyrimidine sites.111 One recent study used exon sequencing of 20000 genes to characterize 14 metastatic melanomas and matched normal DNAs.112 Among 68 genes that appeared to be mutated in the melanomas, the gene GRIN2A encoding an N-methyl-D-aspartate receptor stood out as being mutated in 33% of the samples. In tumor DNA, the proportion of C to T transitions was highly prevalent (>80% of all single base substitutions) and many CC to TT mutations also were observed, again suggesting a role of CPDs in melanoma induction.
Conclusions
Terrestrial solar UV radiation produces various types of DNA photoproducts. The most prevalent ones are CPDs produced in the UVB range of the solar spectrum and also to a low extent by UVA, and oxidized guanines produced predominantly by UVA. Whereas oxidized bases are repaired rapidly and contribute only in a minor way to solar UV mutagenesis, CPDs are persistent lesions and are substantially mutagenic when they contain cytosine or 5-methylcytosine. The currently available data from experimental systems and from skin cancer mutations, including melanoma, overall clearly suggest that CPDs are the most prominent DNA lesion that can initiate melanoma and non-melanoma skin cancers. It will be interesting to examine the mutational landscapes of additional melanoma specimens including primary tumors localized on different body sites to assess the generality of these finding with respect to sunlight and CPD-driven origin of this type of highly malignant tumor.Abbreviations
| UV | Ultraviolet |
| CPD | Cyclobutane pyrimidine dimer |
| (6–4)PP | Pyrimidine (6–4) pyrimidone photoproducts |
| 8-oxo-dG | 8-Oxo-7,8-dihydro-2′-deoxyguanosine |
| XP | Xeroderma pigmentosum |
| NER | Nucleotide excision repair |
| 5mC | 5-Methylcytosine |
Acknowledgements
Work of the authors is supported by the National Institute of Environmental Health Sciences (grant ES 06070 to G. P. P.).References
- D. L. Miller and M. A. Weinstock, Nonmelanoma skin cancer in the United States: incidence, J. Am. Acad. Dermatol., 1994, 30, 774–778 CrossRef CAS.
- A. N. Houghton and D. Polsky, Focus on melanoma, Cancer Cell, 2002, 2, 275–278 CrossRef CAS.
- M. R. Donaldson and B. M. Coldiron, No end in sight: the skin cancer epidemic continues, Semin. Cutaneous Med. Surg., 2011, 30, 3–5 CrossRef CAS.
- M. A. Tucker, Is sunlight important to melanoma causation?, Cancer Epidemiol., Biomarkers Prev., 2008, 17, 467–468 CrossRef.
- R. Greinert, Skin cancer: new markers for better prevention, Pathobiology, 2009, 76, 64–81 CrossRef.
- G. P. Pfeifer, Formation and processing of UV photoproducts: effects of DNA sequence and chromatin environment, Photochem. Photobiol., 1997, 65, 270–283 CrossRef CAS.
- D. L. Mitchell and R. S. Nairn, The biology of the (6–4) photoproduct, Photochem. Photobiol., 1989, 49, 805–819 CrossRef CAS.
- J.-H. Yoon, C.-S. Lee, T. O'Connor, A. Yasui and G. P. Pfeifer, The DNA damage spectrum produced by simulated sunlight, J. Mol. Biol., 2000, 299, 681–693 CrossRef CAS.
- T. Douki, A. Reynaud-Angelin, J. Cadet and E. Sage, Bipyrimidine photoproducts rather than oxidative lesions are the main type of DNA damage involved in the genotoxic effect of solar UVA radiation, Biochemistry, 2003, 42, 9221–9226 CrossRef CAS.
- P. J. Rochette, J. P. Therrien, R. Drouin, D. Perdiz, N. Bastien, E. A. Drobetsky and E. Sage, UVA-induced cyclobutane pyrimidine dimers form predominantly at thymine-thymine dipyrimidines and correlate with the mutation spectrum in rodent cells, Nucleic Acids Res., 2003, 31, 2786–2794 CrossRef CAS.
- A. Besaratinia, T. W. Synold, H. H. Chen, C. Chang, B. Xi, A. D. Riggs and G. P. Pfeifer, DNA lesions induced by UV A1 and B radiation in human cells: comparative analyses in the overall genome and in the p53 tumor suppressor gene, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10058–10063 CrossRef CAS.
- J. Cadet, M. Berger, T. Douki, B. Morin, S. Raoul, J. L. Ravanat and S. Spinelli, Effects of UV and visible radiation on DNA-final base damage, Biol. Chem., 1997, 378, 1275–1286 CAS.
- C. Kielbassa, L. Roza and B. Epe, Wavelength dependence of oxidative DNA damage induced by UV and visible light, Carcinogenesis, 1997, 18, 811–816 CrossRef CAS.
- Z. Kuluncsics, D. Perdiz, E. Brulay, B. Muel and E. Sage, Wavelength dependence of ultraviolet-induced DNA damage distribution: involvement of direct or indirect mechanisms and possible artefacts, J. Photochem. Photobiol., B, 1999, 49, 71–80 CrossRef CAS.
- E. Kvam and R. M. Tyrell, Induction of oxidative DNA base damage in human skin cells by UV and near visible radiation, Carcinogenesis, 1997, 18, 2379–2384 CrossRef CAS.
- X. Zhang, B. S. Rosenstein, Y. Wang, M. Lebwohl, D. M. Mitchell and H. Wei, Induction of 8-oxo-7,8-dihydro-2′-deoxyguanosine by ultraviolet radiation in calf thymus DNA and HeLa cells, Photochem. Photobiol., 1997, 65, 119–124 CrossRef CAS.
- S. Courdavault, C. Baudouin, M. Charveron, A. Favier, J. Cadet and T. Douki, Larger yield of cyclobutane dimers than 8-oxo-7,8-dihydroguanine in the DNA of UVA-irradiated human skin cells, Mutat. Res., Fundam. Mol. Mech. Mutagen., 2004, 556, 135–142 CrossRef CAS.
- S. Mouret, C. Baudouin, M. Charveron, A. Favier, J. Cadet and T. Douki, Cyclobutane pyrimidine dimers are predominant DNA lesions in whole human skin exposed to UVA radiation, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 13765–13770 CrossRef CAS.
- A. Besaratinia, T. W. Synold, B. Xi and G. P. Pfeifer, G-to-T transversions and small tandem base deletions are the hallmark of mutations induced by ultraviolet a radiation in mammalian cells, Biochemistry, 2004, 43, 8169–8177 CrossRef CAS.
- J. Cadet and T. Douki, Oxidatively generated damage to DNA by UVA radiation in cells and human skin, J. Invest. Dermatol., 2011, 131, 1005–1007 CrossRef CAS.
- J. P. Pouget, T. Douki, M. J. Richard and J. Cadet, DNA damage induced in cells by gamma and UVA radiation as measured by HPLC/GC-MS and HPLC-EC and Comet assay, Chem. Res. Toxicol., 2000, 13, 541–549 CrossRef CAS.
- H. Ikehata, K. Kawai, J. Komura, K. Sakatsume, L. Wang, M. Imai, S. Higashi, O. Nikaido, K. Yamamoto, K. Hieda, M. Watanabe, H. Kasai and T. Ono, UVA1 genotoxicity is mediated not by oxidative damage but by cyclobutane pyrimidine dimers in normal mouse skin, J. Invest. Dermatol., 2008, 128, 2289–2296 CrossRef CAS.
- U. P. Kappes, D. Luo, M. Potter, K. Schulmeister and T. M. Runger, Short- and long-wave UV light (UVB and UVA) induce similar mutations in human skin cells, J. Invest. Dermatol., 2006, 126, 667–675 CrossRef CAS.
- A. Besaratinia, S. I. Kim, S. E. Bates and G. P. Pfeifer, Riboflavin activated by ultraviolet A1 irradiation induces oxidative DNA damage-mediated mutations inhibited by vitamin C, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 5953–5958 CrossRef CAS.
- N. S. Agar, G. M. Halliday, R. S. Barnetson, H. N. Ananthaswamy, M. Wheeler and A. M. Jones, The basal layer in human squamous tumors harbors more UVA than UVB fingerprint mutations: a role for UVA in human skin carcinogenesis, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 4954–4959 CrossRef CAS.
- P. C. Hanawalt and A. Sarasin, Cancer-prone hereditary diseases with DNA processing abnormalities, Trends Genet., 1986, 2, 124–129 CrossRef CAS.
- H. N. Ananthaswamy and W. E. Pierceall, Molecular mechanisms of ultraviolet radiation carcinogenesis, Photochem. Photobiol., 1990, 52, 1119–1136 CrossRef CAS.
- G. P. Pfeifer, R. Drouin, A. D. Riggs and G. P. Holmquist, In vivo mapping of a DNA adduct at nucleotide resolution: detection of pyrimidine (6–4) pyrimidone photoproducts by ligation-mediated polymerase chain reaction, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 1374–1378 CrossRef CAS.
- G. P. Pfeifer, R. Drouin, A. D. Riggs and G. P. Holmquist, Binding of transcription factors creates hot spots for UV photoproducts in vivo, Mol. Cell. Biol., 1992, 12, 1798–1804 CAS.
- S. Tommasi, A. B. Oxyzoglou and G. P. Pfeifer, Cell cycle-independent removal of UV-induced pyrimidine dimers from the promoter and the transcription initiation domain of the human CDC2 gene, Nucleic Acids Res., 2000, 28, 3991–3998 CrossRef CAS.
- S. Tornaletti and G. P. Pfeifer, Slow repair of pyrimidine dimers at p53 mutation hotspots in skin cancer, Science, 1994, 263, 1436–1438 CAS.
- Y. Tu, S. Tornaletti and G. P. Pfeifer, DNA repair domains within a human gene: selective repair of sequences near the transcription initiation site, EMBO J., 1996, 15, 675–683 CAS.
- R. Dammann and G. P. Pfeifer, Lack of gene- and strand-specific DNA repair in RNA polymerase III transcribed human tRNA genes, Mol. Cell. Biol., 1997, 17, 219–229 CAS.
- D. E. Brash, J. A. Rudolph, J. A. Simon, A. Lin, G. J. McKenna, H. P. Baden, A. J. Halperin and J. Pontén, A role for sunlight in skin cancer: UV-induced p53 mutations in squamous cell carcinoma, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 10124–10128 CrossRef CAS.
- N. Dumaz, C. Drougard, A. Sarasin and L. Daya-Grosjean, Specific UV-induced mutation spectrum in the p53 gene of skin tumors from DNA-repair-deficient xeroderma pigmentosum patients, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 10529–10533 CrossRef CAS.
- A. Ziegler, D. J. Leffell, S. Kunala, H. W. Sharma, M. Gailani, J. A. Simon, A. J. Halperin, H. P. Baden, P. E. Shapiro, A. E. Bale and D. E. Brash, Mutation hot spots due to sunlight in the p53 gene of nonmelanoma skin cancers, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 4216–4220 CrossRef CAS.
- G. Giglia-Mari and A. Sarasin, TP53 mutations in human skin cancers, Hum. Mutat., 2003, 21, 217–228 CrossRef CAS.
- S. Tommasi, M. F. Denissenko and G. P. Pfeifer, Sunlight induces pyrimidine dimers preferentially at 5-methylcytosine bases, Cancer Res., 1997, 57, 4727–4730 CAS.
- S. Tornaletti and G. P. Pfeifer, Complete and tissue-independent methylation of CpG sites in the p53 gene: implications for mutations in human cancers, Oncogene, 1995, 10, 1493–1499 CAS.
- G. P. Pfeifer, Mutagenesis at methylated CpG sequences, Curr. Top. Microbiol. Immunol., 2006, 301, 259–281 CrossRef CAS.
- A. Sharonov, T. Gustavsson, S. Marguet and D. Markovitsi, Photophysical properties of 5-methylcytidine, Photochem. Photobiol. Sci., 2003, 2, 362–364 CAS.
- Y.-H. You, C. Li and G. P. Pfeifer, Involvement of 5-methylcytosine in sunlight-induced mutagenesis, J. Mol. Biol., 1999, 293, 493–503 CrossRef CAS.
- Y. H. You, D. H. Lee, J. H. Yoon, S. Nakajima, A. Yasui and G. P. Pfeifer, Cyclobutane pyrimidine dimers are responsible for the vast majority of mutations induced by UVB irradiation in mammalian cells, J. Biol. Chem., 2001, 276, 44688–44694 CrossRef CAS.
- H. Ikehata and T. Ono, The mechanisms of UV mutagenesis, J. Radiat. Res., 2011, 52, 115–125 CrossRef CAS.
- D. H. Lee and G. P. Pfeifer, Deamination of 5-methylcytosines within cyclobutane pyrimidine dimers is an important component of UVB mutagenesis, J. Biol. Chem., 2003, 278, 10314–10321 CrossRef CAS.
- R. B. Setlow, The wavelengths in sunlight effective in producing skin cancer: a theoretical analysis, Proc. Natl. Acad. Sci. U. S. A., 1974, 71, 3363–3366 CrossRef CAS.
- G. Kelfkens, F. R. de Gruijl and J. C. van der Leun, Ozone depletion and increase in annual carcinogenic ultraviolet dose, Photochem. Photobiol., 1990, 52, 819–823 CrossRef CAS.
- F. R. de Gruijl, Photocarcinogenesis: UVA vs. UVB radiation, Skin Pharmacol. Physiol., 2002, 15, 316–320 CrossRef CAS.
- F. R. de Gruijl, H. J. Sterenborg, P. D. Forbes, R. E. Davies, C. Cole, G. Kelfkens, H. van Weelden, H. Slaper and J. C. van der Leun, Wavelength dependence of skin cancer induction by ultraviolet irradiation of albino hairless mice, Cancer Res., 1993, 53, 53–60 CAS.
- A. Besaratinia, J. I. Yoon, C. Schroeder, S. E. Bradforth, M. Cockburn and G. P. Pfeifer, Wavelength dependence of ultraviolet radiation-induced DNA damage as determined by laser irradiation suggests that cyclobutane pyrimidine dimers are the principal DNA lesions produced by terrestrial sunlight, FASEB J., 2011 DOI:10.1096/fj.11-187336.
- J. Cadet, T. Douki, J. L. Ravanat and P. Di Mascio, Sensitized formation of oxidatively generated damage to cellular DNA by UVA radiation, Photochem. Photobiol. Sci., 2009, 8, 903–911 CAS.
- J. Cadet, E. Sage and T. Douki, Ultraviolet radiation-mediated damage to cellular DNA, Mutat. Res., 2005, 571, 3–17 CrossRef CAS.
- G. P. Pfeifer, Y. H. You and A. Besaratinia, Mutations induced by ultraviolet light, Mutat. Res., 2005, 571, 19–31 CrossRef CAS.
- R. M. Tyrrell, Role for singlet oxygen in biological effects of ultraviolet A radiation, Methods Enzymol., 2000, 319, 290–296 CAS.
- E. Sage, Distribution and repair of photolesions in DNA: genetic consequences and the role of sequence context, Photochem. Photobiol., 1993, 57, 163–174 CrossRef CAS.
- J. H. Yoon, C. S. Lee, T. R. O'Connor, A. Yasui and G. P. Pfeifer, The DNA damage spectrum produced by simulated sunlight, J. Mol. Biol., 2000, 299, 681–693 CrossRef CAS.
- Y. H. You, D. H. Lee, J. H. Yoon, S. Nakajima, A. Yasui and G. P. Pfeifer, Cyclobutane pyrimidine dimers are responsible for the vast majority of mutations induced by UVB irradiation in mammalian cells, J. Biol. Chem., 2001, 276, 44688–44694 CrossRef CAS.
- C. P. Jacovides, G. P. Gianourakos, D. N. Asimakopoulos and M. D. Steven, Measured Spectra of Solar Ultraviolet Irradiances at Athens Basin, Greece, Theor. Appl. Climatol., 1998, 59, 107–119 CrossRef.
- S. K. Banerjee, R. B. Christensen, C. W. Lawrence and J. E. LeClerc, Frequency and spectrum of mutations produced by a single cis-syn thymine-thymine cyclobutane dimer in a single-stranded vector, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 8141–8145 CrossRef CAS.
- A. Gentil, F. Le Page, A. Margot, C. W. Lawrence, A. Borden and A. Sarasin, Mutagenicity of a unique thymine-thymine dimer or thymine-thymine pyrimidine pyrimidone (6–4) photoproduct in mammalian cells, Nucleic Acids Res., 1996, 24, 1837–1840 CrossRef CAS.
- P. E. M. Gibbs and C. W. Lawrence, U-U and T-T cyclobutane dimers have different mutational properties, Nucleic Acids Res., 1993, 21, 4059–4065 CrossRef CAS.
- J.-S. Taylor and C. L. O'Day, Cis-syn thymine dimers are not absolute blocks to replication by DNA polymerase I of Escherichia coli in vitro, Biochemistry, 1990, 29, 1624–1632 CrossRef CAS.
- M. J. Horsfall, A. Borden and C. W. Lawrence, Mutagenic properties of the T-C cyclobutane dimer, J. Bacteriol., 1997, 179, 2835–2839 CAS.
- B. Vu, V. J. Cannistraro, L. Sun and J. S. Taylor, DNA synthesis past a 5-methylC-containing cis-syn-cyclobutane pyrimidine dimer by yeast pol eta is highly nonmutagenic, Biochemistry, 2006, 45, 9327–9335 CrossRef CAS.
- D. L. Mitchell and A. A. Fernandez, Different types of DNA damage play different roles in the etiology of sunlight-induced melanoma, Pigm. Cell Melanoma Res., 2011, 24, 119–124 CrossRef CAS.
- A. Besaratinia, S. I. Kim and G. P. Pfeifer, Rapid repair of UVA-induced oxidized purines and persistence of UVB-induced dipyrimidine lesions determine the mutagenicity of sunlight in mouse cells, FASEB J., 2008, 22, 2379–2392 CrossRef CAS.
- L. F. Batista, B. Kaina, R. Meneghini and C. F. Menck, How DNA lesions are turned into powerful killing structures: insights from UV-induced apoptosis, Mutat. Res., Rev. Mutat. Res., 2009, 681, 197–208 CrossRef CAS.
- R. E. Johnson, C. M. Kondratick, S. Prakash and L. Prakash, hRAD30 mutations in the variant form of xeroderma pigmentosum, Science, 1999, 285, 263–265 CrossRef CAS.
- R. E. Johnson, S. Prakash and L. Prakash, Efficient bypass of a thymine-thymine dimer by yeast DNA polymerase, Poleta, Science, 1999, 283, 1001–1004 CrossRef CAS.
- C. Masutani, R. Kusumoto, A. Yamada, N. Dohmae, M. Yokoi, M. Yuasa, M. Araki, S. Iwai, K. Takio and F. Hanaoka, The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase eta, Nature, 1999, 399, 700–704 CrossRef CAS.
- J. H. Choi and G. P. Pfeifer, The role of DNA polymerase eta in UV mutational spectra, DNA Repair, 2005, 4, 211–220 CrossRef CAS.
- S. G. Kozmin, Y. I. Pavlov, T. A. Kunkel and E. Sage, Roles of Saccharomyces cerevisiae DNA polymerases Poleta and Polzeta in response to irradiation by simulated sunlight, Nucleic Acids Res., 2003, 31, 4541–4552 CrossRef CAS.
- J. H. Yoon, L. Prakash and S. Prakash, Highly error-free role of DNA polymerase eta in the replicative bypass of UV-induced pyrimidine dimers in mouse and human cells, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 18219–18224 CrossRef CAS.
- Y. Tu, R. Dammann and G. P. Pfeifer, Sequence and time-dependent deamination of cytosine bases in UVB-induced cyclobutane pyrimidine dimers in vivo, J. Mol. Biol., 1998, 284, 297–311 CrossRef CAS.
- N. Jiang and J.-S. Taylor, In vivo evidence that UV-induced C-T mutations at dipyrimidine sites could result from the replicative bypass of cis-syn cyclobutane dimers or their deamination products, Biochemistry, 1993, 32, 472–481 CrossRef CAS.
- Q. Song, V. J. Cannistraro and J. S. Taylor, Rotational position of a 5-methylcytosine-containing cyclobutane pyrimidine dimer in a nucleosome greatly affects its deamination rate, J. Biol. Chem., 2010, 286, 6329–6335 CrossRef.
- F. R. de Gruijl, Skin cancer and solar UV radiation, Eur. J. Cancer, 1999, 35, 2003–2009 CrossRef CAS.
- C. Jhappan, F. P. Noonan and G. Merlino, Ultraviolet radiation and cutaneous malignant melanoma, Oncogene, 2003, 22, 3099–3112 CrossRef CAS.
- B. A. Gilchrest, M. S. Eller, A. C. Geller and M. Yaar, The pathogenesis of melanoma induced by ultraviolet radiation, N. Engl. J. Med., 1999, 340, 1341–1348 CrossRef CAS.
- M. G. Cockburn, J. Zadnick and D. Deapen, Developing epidemic of melanoma in the Hispanic population of California, Cancer, 2006, 106, 1162–1168 CrossRef.
- U. Leiter and C. Garbe, Epidemiology of melanoma and nonmelanoma skin cancer–the role of sunlight, Adv. Exp. Med. Biol., 2008, 624, 89–103 CrossRef.
- L. Pho, D. Grossman and S. A. Leachman, Melanoma genetics: a review of genetic factors and clinical phenotypes in familial melanoma, Curr. Opin. Oncol., 2006, 18, 173–179 CrossRef CAS.
- Z. Tatalovich, J. P. Wilson, T. Mack, Y. Yan and M. Cockburn, The objective assessment of lifetime cumulative ultraviolet exposure for determining melanoma risk, J. Photochem. Photobiol., B, 2006, 85, 198–204 CrossRef CAS.
- I. H. Langford, G. Bentham and A. L. McDonald, Multi-level modelling of geographically aggregated health data: a case study on malignant melanoma mortality and UV exposure in the European Community, Stat. Med., 1998, 17, 41–57 CrossRef CAS.
- J. Moan, A. Dahlback and R. B. Setlow, Epidemiological support for an hypothesis for melanoma induction indicating a role for UVA radiation, Photochem. Photobiol., 1999, 70, 243–247 CrossRef CAS.
- S. Q. Wang, R. Setlow, M. Berwick, D. Polsky, A. A. Marghoob, A. W. Kopf and R. S. Bart, Ultraviolet A and melanoma: a review, J. Am. Acad. Dermatol., 2001, 44, 837–846 CrossRef CAS.
- A. D. Woodhead, R. B. Setlow and M. Tanaka, Environmental factors in nonmelanoma and melanoma skin cancer, J. Epidemiol., 1999, 9, S102–S114 CAS.
- F. R. de Gruijl, H. J. Sterenborg, P. D. Forbes, R. E. Davies, C. Cole, G. Kelfkens, H. van Weelden, H. Slaper and J. C. van der Leun, Wavelength dependence of skin cancer induction by ultraviolet irradiation of albino hairless mice, Cancer Res., 1993, 53, 53–60 CAS.
- F. R. de Gruijl, Photocarcinogenesis: UVA vs. UVB radiation, Skin Pharmacol. Physiol., 2002, 15, 316–320 CrossRef CAS.
- A. de Laat, J. C. van der Leun and F. R. de Gruijl, Carcinogenesis induced by UVA (365-nm) radiation: the dose-time dependence of tumor formation in hairless mice, Carcinogenesis, 1997, 18, 1013–1020 CrossRef CAS.
- P. Autier, J. F. Dore, F. Lejeune, K. F. Koelmel, O. Geffeler, P. Hille, J. P. Cesarini, D. Lienard, A. Liabeuf and M. Joarlette, et al., Cutaneous malignant melanoma and exposure to sunlamps or sunbeds: an EORTC multicenter case-control study in Belgium, France and Germany. EORTC Melanoma Cooperative Group, Int. J. Cancer, 1994, 58, 809–813 CrossRef CAS.
- IARC Working Group, The association of use of sunbeds with cutaneous malignant melanoma and other skin cancers: A systematic review, Int. J. Cancer, 2007, 120, 1116–1122 Search PubMed.
- Y. Jiang, M. Rabbi, M. Kim, C. Ke, W. Lee, R. L. Clark, P. A. Mieczkowski and P. E. Marszalek, UVA generates pyrimidine dimers in DNA directly, Biophys. J., 2009, 96, 1151–1158 CrossRef CAS.
- S. Mouret, C. Philippe, J. Gracia-Chantegrel, A. Banyasz, S. Karpati, D. Markovitsi and T. Douki, UVA-induced cyclobutane pyrimidine dimers in DNA: a direct photochemical mechanism?, Org. Biomol. Chem., 2010, 8, 1706–1711 CAS.
- R. D. Ley, Ultraviolet radiation A-induced precursors of cutaneous melanoma in Monodelphis domestica, Cancer Res., 1997, 57, 3682–3684 CAS.
- R. B. Setlow, E. Grist, K. Thompson and A. D. Woodhead, Wavelengths effective in induction of malignant melanoma, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 6666–6670 CrossRef CAS.
- D. L. Mitchell, A. A. Fernandez, R. S. Nairn, R. Garcia, L. Paniker, D. Trono, H. D. Thames and I. Gimenez-Conti, Ultraviolet A does not induce melanomas in a Xiphophorus hybrid fish model, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 9329–9334 CrossRef CAS.
- E. C. De Fabo, F. P. Noonan, T. Fears and G. Merlino, Ultraviolet B but not ultraviolet A radiation initiates melanoma, Cancer Res., 2004, 64, 6372–6376 CrossRef CAS.
- A. Kamb, D. Shattuck-Eidens, R. Eeles, Q. Liu, N. A. Gruis, W. Ding, C. Hussey, T. Tran, Y. Miki and J. Weaver-Feldhaus, et al., Analysis of the p16 gene (CDKN2) as a candidate for the chromosome 9p melanoma susceptibility locus, Nat. Genet., 1994, 8, 22–26 CrossRef CAS.
- T. Hocker and H. Tsao, Ultraviolet radiation and melanoma: a systematic review and analysis of reported sequence variants, Hum. Mutat., 2007, 28, 578–588 CrossRef CAS.
- P. M. Pollock, F. Yu, L. Qiu, P. G. Parsons and N. K. Hayward, Evidence for u.v. induction of CDKN2 mutations in melanoma cell lines, Oncogene, 1995, 11, 663–668 CAS.
- A. Besaratinia and G. P. Pfeifer, Sunlight ultraviolet irradiation and BRAF V600 mutagenesis in human melanoma, Hum. Mutat., 2008, 29, 983–991 CrossRef CAS.
- N. E. Thomas, M. Berwick and M. Cordeiro-Stone, Could BRAF mutations in melanocytic lesions arise from DNA damage induced by ultraviolet radiation?, J. Invest. Dermatol., 2006, 126, 1693–1696 CrossRef CAS.
- C. D. Van Raamsdonk, K. G. Griewank, M. B. Crosby, M. C. Garrido, S. Vemula, T. Wiesner, A. C. Obenauf, W. Wackernagel, G. Green, N. Bouvier, M. M. Sozen, G. Baimukanova, R. Roy, A. Heguy, I. Dolgalev, R. Khanin, K. Busam, M. R. Speicher, J. O'Brien and B. C. Bastian, Mutations in GNA11 in uveal melanoma, N. Engl. J. Med., 2010, 363, 2191–2199 CrossRef CAS.
- A. Besaratinia and G. P. Pfeifer, Uveal melanoma and GNA11 mutations: a new piece added to the puzzle, Pigm. Cell Melanoma Res., 2011, 24, 18–20 CrossRef CAS.
- Z. A. Abdel-Malek, A. L. Kadekaro and V. B. Swope, Stepping up melanocytes to the challenge of UV exposure, Pigm. Cell Melanoma Res., 2010, 23, 171–186 CrossRef CAS.
- G. G. McGill, M. Horstmann, H. R. Widlund, J. Du, G. Motyckova, E. K. Nishimura, Y. L. Lin, S. Ramaswamy, W. Avery, H. F. Ding, S. A. Jordan, I. J. Jackson, S. J. Korsmeyer, T. R. Golub and D. E. Fisher, Bcl2 regulation by the melanocyte master regulator Mitf modulates lineage survival and melanoma cell viability, Cell, 2002, 109, 707–718 CrossRef CAS.
- E. D. Pleasance, R. K. Cheetham, P. J. Stephens, D. J. McBride, S. J. Humphray, C. D. Greenman, I. Varela, M. L. Lin, G. R. Ordonez, G. R. Bignell, K. Ye, J. Alipaz, M. J. Bauer, D. Beare, A. Butler, R. J. Carter, L. Chen, A. J. Cox, S. Edkins, P. I. Kokko-Gonzales, N. A. Gormley, R. J. Grocock, C. D. Haudenschild, M. M. Hims, T. James, M. Jia, Z. Kingsbury, C. Leroy, J. Marshall, A. Menzies, L. J. Mudie, Z. Ning, T. Royce, O. B. Schulz-Trieglaff, A. Spiridou, L. A. Stebbings, L. Szajkowski, J. Teague, D. Williamson, L. Chin, M. T. Ross, P. J. Campbell, D. R. Bentley, P. A. Futreal and M. R. Stratton, A comprehensive catalogue of somatic mutations from a human cancer genome, Nature, 2009, 463, 191–196 CrossRef.
- G. P. Pfeifer, Environmental exposures and mutational patterns of cancer genomes, Genome Med., 2010, 2, 54 CrossRef.
- I. Mellon, G. Spivak and P. C. Hanawalt, Selective removal of transcription-blocking DNA damage from the transcribed strand of the mammalian DHFR gene, Cell, 1987, 51, 241–249 CrossRef CAS.
- C. Greenman, P. Stephens, R. Smith, G. L. Dalgliesh, C. Hunter, G. Bignell, H. Davies, J. Teague, A. Butler, C. Stevens, S. Edkins, S. O'Meara, I. Vastrik, E. E. Schmidt, T. Avis, S. Barthorpe, G. Bhamra, G. Buck, B. Choudhury, J. Clements, J. Cole, E. Dicks, S. Forbes, K. Gray, K. Halliday, R. Harrison, K. Hills, J. Hinton, A. Jenkinson, D. Jones, A. Menzies, T. Mironenko, J. Perry, K. Raine, D. Richardson, R. Shepherd, A. Small, C. Tofts, J. Varian, T. Webb, S. West, S. Widaa, A. Yates, D. P. Cahill, D. N. Louis, P. Goldstraw, A. G. Nicholson, F. Brasseur, L. Looijenga, B. L. Weber, Y. E. Chiew, A. DeFazio, M. F. Greaves, A. R. Green, P. Campbell, E. Birney, D. F. Easton, G. Chenevix-Trench, M. H. Tan, S. K. Khoo, B. T. Teh, S. T. Yuen, S. Y. Leung, R. Wooster, P. A. Futreal and M. R. Stratton, Patterns of somatic mutation in human cancer genomes, Nature, 2007, 446, 153–158 CrossRef CAS.
- X. Wei, V. Walia, J. C. Lin, J. K. Teer, T. D. Prickett, J. Gartner, S. Davis, K. Stemke-Hale, M. A. Davies, J. E. Gershenwald, W. Robinson, S. Robinson, S. A. Rosenberg and Y. Samuels, Exome sequencing identifies GRIN2A as frequently mutated in melanoma, Nat. Genet., 2011, 43, 442–446 CrossRef CAS.
Footnote |
| † Contribution to the themed issue on the biology of UVA. |
| This journal is © The Royal Society of Chemistry and Owner Societies 2012 |