DNA damage spectra induced by photosensitization†
Bernd
Epe
*
Institute of Pharmacy and Biochemistry, University of Mainz, Staudingerweg 5, D-55099, Mainz, Germany. E-mail: epe@uni-mainz.de; Fax: +49-6131-39 25521; Tel: +49-6131-39 24309
First published on 8th September 2011
Abstract
DNA damage induced by photosensitization is not only responsible for the genotoxic effects of various types of drugs in the presence of light, but is also relevant for some of the adverse effects of sunlight, in particular in the UVA and visible range of the spectrum. The types of DNA modifications induced are very diverse and include pyrimidine dimers, covalent adducts, various base modifications generated by oxidation, single-strand breaks and (regular and oxidized) sites of base loss. The ratios in which the various modifications are formed (damage spectra) can be regarded as a fingerprint of the damaging mechanism. Here, we describe the damage spectra of various classes of photosensitizers in relation to the underlying damaging mechanisms. In mammalian cells irradiated with solar radiation, damage at wavelengths <400 nm is characteristic for a (not yet identified) endogenous type-I or type-II photosensitizer. In the UVA range, however, both direct DNA excitation and photosensitized damage appear to be relevant, and there are indications that other chromophore(s) are involved than in the visible range.
 Bernd Epe | Bernd Epe obtained his PhD (organic chemistry) at the University of Kiel in 1977. He was post-doc at the Max-Planck-Institute for Molecular Genetics, Berlin, and habilitated at the Institute of Pharmacology and Toxicology of the University of Würzburg in 1989. Since 1994, he has been full Professor of Pharmacology and Toxicology at the Institute of Pharmacy and Biochemistry of the University of Mainz. His main research activities deal with oxidative DNA damage in mammalian cells, its repair and its consequences. |
1. Introduction
When an organic molecule absorbs light, the excited state generated is in most cases too short-lived to react chemically with other molecules, and the excitation energy is thermally dissipated. In some cases, however, the lifetimes are long enough to allow chemical reactions, often because the initially generated excited singlet state is converted into a more stable triplet state by so-called intersystem crossing. These compounds are called photoreactive. If after reaction the absorbing molecule is ultimately re-generated in its ground state, i.e. if it acts as a catalyst, it is called a photosensitizer. If the absorbing chromophore is destroyed in the photoreaction, this is called photobleaching.In cells, photoreactive compounds can react with many different targets. Therefore, it can be assumed that all photoreactive compounds are phototoxic. Because the excited molecules are generally radicals, membrane lipids with unsaturated fatty acid residues are preferential reaction partners, especially for compounds with low hydrophilicity. DNA is quantitatively only a minor target in many cases. However, its reaction gives rise to DNA modifications and, subsequently, mutations. Therefore, the reaction of excited photosensitizers with DNA is of high relevance, even in cases in which the phototoxicity is predominantly mediated by other target molecules.1 The ratio between photogenotoxicity (caused by a reaction with DNA) and phototoxicity (which can result from reactions with all cellular targets) can differ largely, depending on the nature of the photoreactive compound, i.e. its hydrophilicity, its affinity to DNA (non-covalent binding) and its photochemical reactivity. However, genotoxicity may never be completely absent since the reactivity of excited photosensitizers and other photoreactive compounds is generally high enough to react with DNA and since reactive oxygen species (ROS) and other primary products formed by photosensitization can give rise to DNA modifications in secondary reactions (see below).
Mechanistically, DNA damage induction by photosensitizers and other photoreactive substances can be classified into (1) direct reactions of the excited molecules with DNA, (2) reactions mediated by ROS and (3) reactions involving other secondary intermediates, for example decomposition products of the excited molecules or lipid peroxidation products. All three types of reaction can be further subdivided, as indicated in Table 1. While some DNA modifications (e.g. cyclobutane pyrimidine dimers) are specifically generated by only one type of reaction mechanism, other DNA lesions such as many base modifications formed by oxidation, sites of base loss (AP sites) and single-strand breaks (SSB) are produced to some extent by virtually all types of DNA damaging photoreactive compounds (Fig. 1). Nevertheless, the ratio in which the various lesions are generated (damage spectrum) is often a characteristic fingerprint of the damaging mechanism. This is particularly expected for those photosensitizers which damage DNA indirectly via ROS, but conceivably also for some type-I photosensitizers.
| Type of DNA damage | Agents (examples) | Major DNA modifications |
|---|---|---|
| 1. Direct damage | ||
| covalent binding | psoralens | photoadducts |
| energy transfer | acetophenone | cyclobutane pyrimidine dimers |
| one-electron oxidation | type-I-photosensitizers | oxidised DNA bases |
| 2. ROS-mediated damage | ||
| via singlet oxygen | porphyrins | oxidised guanines |
| viahydroxyl radicals | extracell. ROS generation | SSB, sites of base loss, oxidised DNA bases |
| 3. Damage mediated by secondary products other than ROS | ||
| viadecomposition products | chlorophenothiazines | adducts, SSB, sites of base loss, oxidised DNA bases |
| vialipid peroxides | adducts | |
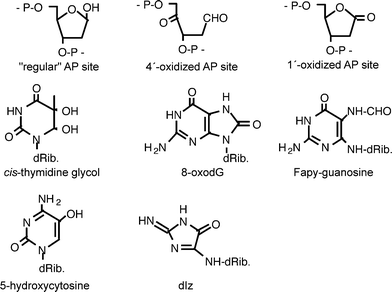 | ||
| Fig. 1 Chemical structures of some DNA modifications induced by photosensitization. | ||
The consequences of DNA damage induced by photosensitizers and photoreactive compounds, in particular cytotoxicity, induction of mutations and initiation of carcinogenesis, obviously depend on the spectrum of DNA modifications induced. Defined types of damage spectra (characteristic for the underlying reaction mechanisms and classes of photosensitizers) should often result in defined, substance-independent consequences.
Here, an overview is given about the DNA damage spectra induced by photosensitization, structured according to the reaction mechanisms indicated in Table 1. In particular, data obtained from the characterization of the damage spectrum by means of repair enzymes as probes are combined with results from more specific, albeit often less comprehensive, analyses by other techniques and with results of mutation analyses, which often allow conclusions with regard to the underlying DNA modifications.
2. Methods for the analysis of DNA damage spectra
Techniques available to analyse the DNA damage generated by photosensitization have been improved considerably in recent years and were described in comprehensive reviews.2,3HPLC coupled with tandem mass spectrometry (HPLC/MS-MS) now allows to quantify both the different forms of pyrimidine dimers and various types of base modifications generated by oxidation in cellular DNA at biologically relevant doses. 8-Oxo-7,8-dihydroguanine (8-oxoGua), which is generated by most oxidants in relatively high yield, can also be determined with similar sensitivity by HPLC coupled with an electrochemical detector (HPLC/ECD) and is often used as a marker lesion for all kinds of oxidative stress in mammalian cells. This is not always straightforward, however, since at low levels of damage an artifactual generation of 8-oxoGua during isolation and hydrolysis of DNA has to be taken into consideration,4 while at high doses of an oxidant 8-oxoGua is easily further oxidized to secondary products.5 Determination of 8-oxoGua and some other base lesions with antibodies is also feasible, but quantitative analyses are rarely conducted.6,7The alkaline elution technique, alkaline unwinding and the comet assay allow a very sensitive quantification of SSB in cellular mammalian DNA. In combination with repair enzymes as probes (Table 1), the methods can be used to determine various other types of DNA damage (cyclobutane pyrimidine dimers, uracil, base modifications generated by oxidation, sites of base loss) with the same sensitivity and without DNA hydrolysis. Thus, different classes of lesions can be quantified in parallel.8–11 Shortcomings of these techniques are the limited and not always fully established substrate specificity of the enzymatic probes and the need for calibration with a reference damage.
3. Direct DNA damage
3.1 Covalent binding
Photoexcited compounds can react directly with DNA either by covalent binding (resulting in adduct formation), by triplet–triplet energy transfer and by one-electron transfer or hydrogen abstraction. The best-studied examples for the first type of reaction are psoralens (furocoumarins), which intercalate into DNA and react by a (photochemically allowed) [2 + 2] cycloaddition between a furocoumarin double bond and the 5,6-double bond of a thymine or cytosine.12 Several of the furocoumarins are bifunctional and can give rise to DNA cross-links. In vivo, this adduct formation and the resulting phototoxicity are exploited in the so-called PUVA therapy to kill highly proliferating cells in the skin. Structurally related natural products, e.g. the alkaloid dictamnine, give rise to similar adducts.13 It should be noted, however, that other types of photodamage to DNA can often take place in parallel to photobinding, e.g. photooxidation by singlet oxygen.143.2 Triplet–triplet energy transfer
The second type of direct DNA damage by photoexcited compounds, triplet–triplet energy transfer, is energetically only possible if the triplet energy of the photoexcited compound is higher than the triplet energy of thymine in DNA, which is the lowest one of the four DNA bases. The exact triplet energy levels of thymine residues depend on the sequence context. However, the minimum triplet energy of a photosensitizer to allow this type of reaction appears be around 270 kJ mol−1.15 This condition is fulfilled by several photosensitizers absorbing in the UVA range, e.g.acetophenone, pyridopsoralens,16 fluoroquinolones17 and some non-steroidal anti-inflammatory drugs.18 The energy transfer to DNA appears to result rather selectively in the formation of cyclobutane thymine dimers. This is in striking contrast to the direct excitation of DNA by UVC or UVB, which not only generates cyclobutane dimers involving cytosines in relatively high yields in addition to thymine dimers, but also gives rise to so-called (6–4) photoproducts.3 Although the photophysical and photochemical processes in the DNA are complex and difficult to analyse, it appears possible that excited singlet states of DNA bases are required to generate the (6–4) photoproducts and that an excitation of cytosines, which in most cases cannot take place by energy transfer, is responsible for most of the cytosine dimers and mixed cytosine-thymine dimers generated by direct excitation of DNA.19It is interesting to note that there is a weak absorption of DNA also in the UVA range of the spectrum, which may be explained by the delocalization of excited states over several bases (excitons).20,21 Excitation of DNA in this range also results in a rather selective formation of cyclobutane thymine dimers and no (6–4) products, i.e. in a damage spectrum very similar to that generated by energy transfer.
An analysis of the DNA damage spectrum generated by acetophenone as a photosensitizer under irradiation with a 333 nm argon ion laser is shown in Fig. 2, left panel. In this analysis, DNA modifications recognized by various repair enzymes (see Table 2) were quantified in parallel with SSB by means of a relaxation assay using supercoiled DNA from bacteriophage PM2 (PM2 DNA) as a target.8,22 The results indicate that cyclobutane pyrimidine dimers recognized by UV endonuclease from Micrococcus luteus were generated in approximately 10-fold higher yields than oxidatively formed purine modifications recognised by Fpg protein and 100-fold higher yields than sites of base loss (recognized by endonuclease IV and exonuclease III) and SSB. Pyrimidine modifications generated by oxidation (sensitive to endonuclease III) were also very rare. The DNA damage by the irradiation alone at this wavelength (333 nm) was much lower than the photosensitized damage (400-fold and 700-fold in the case of Fpg-sensitive modifications and cyclobutane pyrimidine dimers, respectively), but still detectable (Fig. 2, right panel). Surprisingly, the damage spectrum without photosensitizer is remarkably similar to that in the presence of acetophenone. These results nicely complement the DNA damage analysis of the different types of pyrimidine dimers. The selectivity of the damage generated by photoexcited ketones can be exploited to generate cyclobutane thymine dimers in oligonucleotides on a preparative scale.23
| Repair enzyme | Base modifications | AP sitesb | ||||
|---|---|---|---|---|---|---|
| CPD c | 8-oxoGua d | FapyGe | FapyAf | oxPyrg | ||
| a For reviews, see ref. 72–75. b 4′- and 1′-oxidized AP sites are very well recognized by endonuclease IV and APN1, but poor substrates for the other enzymes.45 c Cyclobutane pyrimidine dimers. d 8-Oxo-7,8-dihydroguanine. e 2,6-Diamino-4-hydroxy-5-formamidopyrimidine. f 4,6-Diamino-5-formamidopyrimidine. g 5,6-Dihydropyrimidines and other oxidized pyrimidines. | ||||||
| Fpg protein (E. coli) | — | + | + | + | — | + |
| Endonuclease III (E. coli) | — | — | — | + | + | + |
| OGG1 (S. cerevisiae) | — | + | + | — | — | + |
| NTG1 (S. cerevisiae) | — | — | + | + | + | + |
| T4endoV (bacteriophage) | + | — | — | — | — | + |
| UV endonuclease (M. luteus) | + | — | — | — | — | + |
| Endonuclease IV (E. coli) | — | — | — | — | — | + |
| APN1 (S. cerevisiae) | — | — | — | — | — | + |
| Exonuclease III (E. coli) | — | — | — | — | — | + |
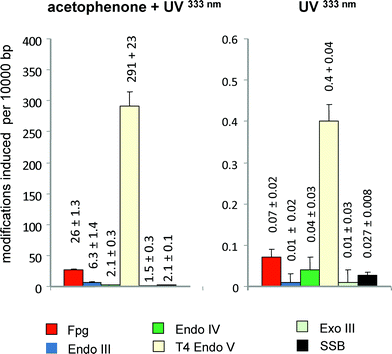 | ||
| Fig. 2 Spectrum of DNA modifications generated per J mm−2 by acetophenenone (6 mM) plus UVA (333 nm) (left panel) or UV (330 nm) alone (right panel) in cell-free DNA. Columns indicate the numbers of SSB and modifications recognized by various repair enzymes (see Table 1) after irradiation of supercoiled PM2 DNA with the 333 nm band of an Argon laser (2.9 kW m−2) in phosphate buffer (pH7.4). Data are taken from Epe et al. (1993).22 | ||
3.3 Type-I reactions
The third type of damage generation in a direct reaction between an excited photosensitizer and DNA is the so-called type-I reaction. In most cases, a hydrogen atom or an electron is abstracted by the triplet-excited state, which is a free radical by nature. Guanine has the lowest one-electron oxidation potential of the DNA bases,24 and therefore DNA modifications result predominantly from oxidation of this base. The ionization potential is particularly low if the guanine is flanked by another purine on its 3′ side.25–27 The reactions of the guanine radicals and the formation of stable products have been studied in great detail (for reviews, see ref. 5 & 28). Further (one-electron) oxidation of the guanine radical appears to result in the generation of 8-oxoGua in the DNA as the most frequent modification. The modification causes characteristic GC to TA transversions during replication.29–31 It is also formed in DNA by hydroxyl radicals and many other oxidants, e.g. singlet oxygen (see below) and bromate32 and is the most frequently used biomarker for oxidative stress at the DNA.In principle, the primarily formed guanine radicals can stabilize in many other ways. Thus, further oxidation (e.g. by superoxide) can result in the formation of 2,5-diamino-4H-imidazol-4-one residues (dIz in Fig. 1) by complicated rearrangements, which hydrolyse to 2,2,4-triamino-5(2H)-oxazolones. The yields of these products appear to be much smaller in DNA than in isolated nucleosides, but may still be comparable to those of 8-oxoGua even at doses at which the product distribution is not influenced by secondary oxidation of 8-oxoGua.33 As an alternative to oxidation, the guanine radicals formed in the primary reaction can stabilize by one-electron reduction following hydration and opening of the imidazole ring, giving rise to the formation of 2,6-diamino-4-hydroxy-5-formamidopyrimidine (Fapy-G) residues.34 Therefore, it may be expected that the final product spectrum of type-I reactions depends both on the redox potential of the exited photosensitizer (which should influence the selectivity of the primary attack) and the redox environment at the DNA (which should influence the stabilization step of the DNA radical).
A classical reference photosensitizer that modifies DNA by a type-I mechanism is riboflavin.25 A DNA damage spectrum obtained by means of various repair endonucleases (Fig. 3, left panel) indicates a very high selectivity for base damage at guanine residues, i.e. a high number of sites sensitive OGG1 and Fpg compared to SSB and sites sensitive to endonuclease III and endonuclease IV. However, only 20% of the Fpg-sensitive guanine modifications are 8-oxoGua according to analysis by HPLC with an electrochemical detector. The remaining base modifications are sensitive to the repair glycosylase NTG1 and are most probably responsible for the high number of GC to CG mutations observed when DNA modified under the same conditions in transfected into bacteria.35 Most of the mutated guanine residues are positioned 5′ to another purine, in accordance with the assumption that the guanines were oxidised in type-I reactions (see above). Although a chemical identification of the NTG1-sensitive sites generated by photoexcited riboflavin has not yet been carried out, 2,5-diamino-4H-imidazol-4-one residues would be good candidate lesions, in particular since they were shown to cause GC to CG mutations.36,37
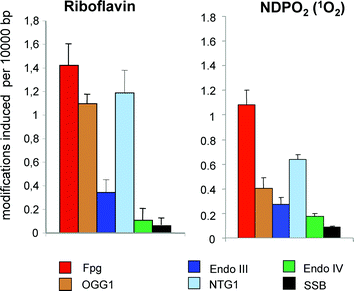 | ||
| Fig. 3 Spectrum of DNA modifications generated by riboflavin (30 μM) plus visible light (1 min) (left panel) or chemically generated singlet oxygen in cell-free PM2 DNA (right panel). Columns indicate the numbers of SSB and modifications recognized by various repair enzymes (see Table 1). Visible light was from a halogen lamp (1000 W, 50 cm distance). Singlet oxygen was generated by thermal decomposition (2 h at 37 °C) of 1 mM NDPO2 in a D2O buffer. Data are taken from Schulz et al. (2000).35 | ||
The selective oxidation of guanine residues has been observed for many other type-I photosensitizers. Mechanistically interesting examples are viologen linked pyrene conjugates, in which a photoinduced intramolecular electron transfer mechanism strongly favors a type-I reaction with DNA.38 On the other hand, some reactive type-I photosensitizers appear to attack the sugar moiety of DNA as well. An instructive example is the β-carboline alkaloid harmine. The compound has an absorption maximum in the UVA range (∼360 nm) and was previously shown to generate SSB when irradiated in the presence of DNA by a type-I mechanism.39 The DNA damage spectrum (Fig. 4) differs substantially from that induced by photoexcited riboflavin (Fig. 3, left panel) and reveals the generation of a relatively high number of - probably oxidized - sites of base loss sensitive to endonuclease IV. In addition, energy transfer to DNA takes place, as indicated by the presence of sites sensitive to T4 endonuclease V, but not to endonuclease IV. (The number of sites detected by the combined activities of the two enzymes is higher than the number of sites of base loss dected by endonuclease IV alone.)
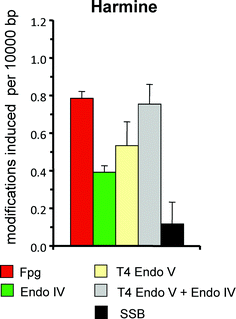 | ||
| Fig. 4 Spectrum of DNA modifications generated harmine (20 μM) plus UVA (360 nm; 24 kJ m−2) in PM2 DNA in phosphate buffer at pH 7.4. Columns indicate the numbers of SSB and modifications recognized by various repair enzymes (see Table 1). Data are taken from Gonzalez et al., submitted. | ||
4. DNA damage mediated by ROS
Photoexcited compounds can react with molecular oxygen in so-called type-II reactions either by electron transfer or by energy transfer.40 The product in the former case is the superoxide ion, which does not react with DNA directly. However, within cells it may give rise to the formation of highly reactive hydroxyl radicals in a metal-catalysed Haber–Weiss reaction. The alternative reaction, the triplet–triplet energy transfer from the excited photosensitizer to oxygen, generates singlet oxygen, which is able to directly damage DNA, although it is much less reactive than hydroxyl radicals. This latter type-II reaction is often dominating and responsible for the DNA damage by many photoreactive drugs absorbing at wavelengths >360 nm.4.1 Hydroxyl radicals
The DNA damage spectrum generated by hydroxyl radicals as well as the underlying reaction mechanisms have been analysed in great detail, in particular because hydroxyl radicals are responsible for most of the DNA damage generated by ionizing radiation. Interestingly, even low-LET ionizing radiation generates much higher yields of clustered (i.e. closely opposed or adjacent) modifications than expected for an independent damage formation. This includes not only double-strand breaks, but also base modifications and/or AP sites.41 In agreement with the high reactivity and therefore low selectivity of hydroxyl radicals, many types of DNA modifications are generated. According to the quantification of cellular DNA base damage induced by ionizing radiation using the HPLC-MS/MS technique, oxidation products of pyrimidines such as thymine glycol (5,6-dihydroxy-5,6-dihydrothymine) and purines such as 8-oxoGua and Fapy-G (2,6-diamino-4-hydroxy-formamidopyrimidine) are generated in approximately similar yields.42,43 The absolute number of thymine glycols determined in these studies (97 lesions per 109 bases per Gy) on the other hand equals the number of SSB (83 lesions per 109 bases per Gy) determined by gradient centrifugation by Kohn et al. in 1976.44 The damage analysis with repair enzymes as probes basically confirm these results and indicate in addition that sites of base loss are only slightly less frequent than SSB (Fig. 5).45,46 As expected, the damage spectrum generated by superoxide in the presence of an iron complex (Fe(III)-EDTA) closely resembles that generated by ionizing radiation, at least in cell-free DNA.47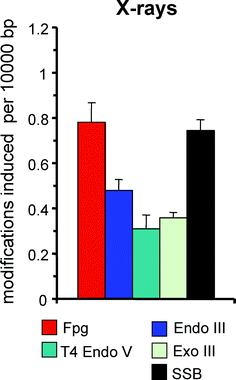 | ||
| Fig. 5 Spectrum of DNA modifications generated by ionizing radiation (20 Gy) in PM2 DNA in phosphate buffer at pH 7.4. Columns indicate the numbers of SSB and modifications recognized by various repair enzymes (see Table 1). Data are taken from Epe et al. (1996).46 | ||
Free hydroxyl radicals can also be generated by photolysis of compounds such as N-hydroxypyridinethiones46 and furocumarin hydroperoxides,47 and the damage spectra observed after irradiation of the these compounds in cells or with cell-free DNA are similar to those caused by ionizing radiation. It is interesting to note that tert-butoxyl radicals, which can be generated by photodecomposition of the water soluble peroxy ester [4-(tert-butyldioxycarbonyl)benzyl]triethylammonium chloride (BCBT), are much more selective than hydroxyl radicals and seem to generate quite specifically 8-oxoGua, as judged from the damage analysis by repair enzymes and from the mutation spectrum, which consists virtually exclusively of GC to TA transversions, the signature mutation of 8-oxoGua.35,48
4.2 DNA damage by singlet oxygen
There is little doubt that the primary reaction between singlet oxygen and DNA gives rise to two unstable diastereomeric 4,8-endoperoxides of guanine residues by [4 + 2] cycloadditon (Diels–Alder reaction). Surprisingly, the stable products generated from these endoperoxides are quite different in isolated nucleosides and in DNA. In the former case, a spiroiminodihydantoine is formed by rearrangementvia a 5-hydroxy-8-oxoguanine intermediate.5,49,50 In DNA and in cells, however, 8-oxoGua was found as the major product by HPLC-MS-MS analysis using a chemical source of singlet oxygen.51 The generation of 8-oxoGua from the initially formed cycloaddition products requires a reduction step, which is mechanistically not yet understood.The analysis by repair enzymes confirms the high selectivity of singlet oxygen for guanine residues.52,53 After damage of isolated DNA with chemically generated singlet oxygen under conditions at which a secondary oxidation of primary DNA modifications is very unlikely, oxidised pyrimidines (sensitive to endonuclease III), sites of base loss (sensitive to endonuclease IV) and SSB were found to be generated in only low yields (Fig. 3, right panel). Surprisingly, however, more than 50% of the purine modifications appear to be other lesions than 8-oxoGua, since they were recognized by Fpg and NTG1, but not by OGG1.35 This notion was confirmed by the mutation spectrum that was obtained when the damaged DNA was transfected into bacteria. Mutations were exclusively observed at GC pairs, but the signature mutations for 8-oxoGua, namely GC to TA transversions, was found in only 50% of the cases, the rest being GC to CG transversions.35 It has to be concluded that unidentified guanine modifications, possibly 2,5-diamino-4H-imidazol-4-one, contribute significantly to the DNA damage spectrum generated by pure singlet oxygen. High, but not exclusive formation of GC to TA transversions was also reported for several photosensitizers that damage DNA predominantly via type-II reaction, e.g. the benzo[a]quinolizine derivative Ro 19-802254 and, in yeast, the fluoroquinolone rufloxacin.55 A preference for guanine residues positioned on the 5′ side of another purine, as characteristic for the one-electron oxidation by type-I photosensitizers, is not observed with singlet oxygen.
5. DNA damage by decomposition products and lipid peroxides
Photodecomposition is frequently observed during irradiation of photoreactive drugs. In some cases, reactive radicals are generated from the parent compounds that can either bind covalently to DNA or give rise to oxidative damage to DNAviaelectron transfer mechanisms. A well-studied example is the phenothiazine chloropromazine.56 At least part of the photogenotoxicty of the compound probably results from covalent binding of the dehalogenated aromatic ring system to the C-8 position of guanine.57 The relative yields of covalent adducts on the one hand and modifications generated by oxidation on the other remain to be established.In some cases, the photodecomposition directly generates ROS. Examples for this type of reaction are N-hydroxypyridinethiones46 and furocumarin hydroperoxides47 and BCBT,48 described in chapter 4.1.
As pointed out in the Introduction, lipids in cell membranes are most likely preferential targets for lipophilic photosensitizers.58 Products of lipid peroxidation, in particular unsaturated aldehydes, are well known to generate DNA adducts.59–62 However, the contribution of this type of indirect DNA damage to the damage spectra and photogenotoxicity of photosensitizers is largely unknown.
6. DNA damage mediated by endogenous photosensitizers
While the cytotoxic and genotoxic effects of UVB in mammalian cells are well explained by the direct excitation of DNA and the resulting very efficient generation of pyrimidine dimers, the chromophores responsible for the DNA damage generated at longer wavelengths are less clear. The action spectrum (wavelength dependence) for the induction of pyrimidine dimers extends far into the UVA range.63–65 Because of the absence of (6–4) photoproducts and dominance of thymine dimers observed after irradiation with UVA, it is tempting to speculate that an energy transfer from unidentified cellular photosensitizers is responsible for this type of damage (see chapter 3.2). However, recent data suggest that a very weak direct absorption of DNA in this wavelength range could also explain the findings.20,21 The origin of the cellular pyrimidine dimers generated by UVA therefore remains to be established.The action spectrum observed for the formation of purine modifications sensitive of Fpg protein (caused by oxidation) clearly deviates from that of pyrimidine dimers. In the UVA range, even at 360 nm, the yield of cyclobutane pyrimidine dimers is still higher than that of Fpg-sensitive sites (Fig. 6, left panel),63,65,66 but at wavelengths >400 nm the formation of oxidised purines is dominating, having a maximum around 420 nm and extending well beyond 450 nm. The DNA damage spectrum at these wavelengths is characteristic for type-I and type-II photosensitizers such as hematoporphrin or riboflavin, i.e. not only pyrimidine dimers, but also single-strand breaks, sites of base loss and oxidised pyrimidines (sensitive to endonuclease III) are absent or generated in very low yields.67 The endogenous photosensitizer responsible for this damage has not yet been identified, but many chromophores in mammalian cells have been considered as photobiologically active.68 Many porphyrins have maximum absorption around 420 nm. When the cellular porphyrin synthesis was increased by pretreatment with 5-aminolaevulinic acid (5-ALA), the number of visible-light induced Fpg-sensitve lesions was four-fold increased.67 However, the concentration of cellular porphyrin under these conditions was raised by a factor of 17. Moreover, at the same level of Fpg-sensitive sites induced by visible light, the associated cytotoxicity and also the number of SSB was clearly higher in the cells pretreated with ALA than in untreated cells. It has to be concluded that either protoporphyrin (induced by 5-ALA) is quite different from the endogenous photosensitizer responsible for the damage in untreated cells, or the cellular localization of the excess of porphyrins in the 5-ALA-pretreated cells is different from that of the basal levels of porphyrins. It is also interesting to not that the endogenous chromophore responsible for the oxidative base damage at wavelengths >400 nm appears to be destroyed under irradiation (photobleaching) since DNA damage generation under continuous irradiation is saturated at low levels (0.3 Fpg-sensitive modifications per 106 bp in HaCat cells).67 In contrast, no such saturation was observed for the generation of 8-oxoGua by UVA.66 This might be an indication that the chromophores responsible for the oxidative base damage by visible light and UVA are different.
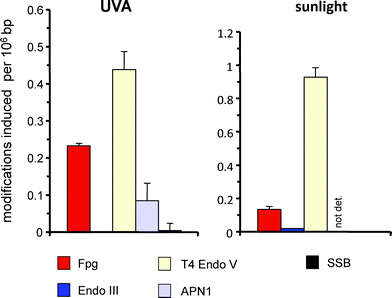 | ||
| Fig. 6 Spectrum of DNA modifications induced by UVA (360 nm; 12 kJ m−2) in primary human skin fibroblasts (left panel) and by natural sunlight (4 min; 88000 lux) in HaCat cells. Columns indicate the numbers of SSB and modifications recognized by various repair enzymes (see Table 1). Data are taken from Eiberger et al. (2008)76 (UVA) and Pflaum et al. (1998)67 (natural sunlight). | ||
The consequences and biological relevance of the DNA damage induced by endogenous photosensitizers remain to be established. When mammalian cells (e.g. primary keratinocytes, HaCat cells or L1210 cells), are irradiated with low doses of natural sunlight, the yield of oxidatively generated purine modifications sensitive to Fpg protein is approx. seven to ten-fold lower than that of pyrimidine dimers (Fig. 6, right panel)67 and virtually all of the oxidative base damage is induced in the UVA and visible range. With respect to the potency to induce mutations in AS52 cells and also micronuclei in melanoma cells, pyrimidine dimers generated by UVB and Fpg-sensitive base modifications generated by photosensitization appear to be equally effective.69,70 With respect to the DNA damage generation by solar light in human skin, it has to be considered that the longer wavelengths responsible for the oxidative base damage have a deeper penetration than the shorter wavelengths responsible for the generation of pyrimidine dimers. Moreover, a photobleaching of the chromophore responsible for the oxidative base damage67 means that experiments at high dose rates might easily cause an underestimation of the photosensitizer-mediated effects if extrapolated to the low dose rates that are often characteristic for human exposure to sunlight.
With respect to the role of endogenous photosensitizers for human skin cancer, it is often assumed that effects on the level of tumor promotion (gene expression, cell cycle regulation) are more important than the generation of DNA damage and mutations (tumor initiation). Thus, the activation of MAPK pathways (p38, JNK) and, in consequence, transcription factors such as AP-1 by photosensitized generation of ROS is well established and can easily explain tumor promotion.71 The actual contribution of the genotoxic effects of endogenous photosensitizers in comparison to the various influences on signal transduction and cell division is expected to depend on the doses and dose rates. Further experiments, e.g. with mice deficient in the repair of DNA damage generated by oxidation, will help to better understand the relevance of DNA damage induced by photosensitization for the different types of skin cancer.
7. Conclusions
Photosensitizers and other photoreactive compounds can give rise to a very broad spectrum of DNA modifications. These cause mutations and other genotoxic effects, but can also be exploited to selectively kill cells by irradiation with UVA or visible light. A comprehensive analysis of the DNA damage spectra in relation to the chemical structures and photophysical characteristics of the underlying chromophores will help to design better photosensitizers for therapeutic applications and to assess the photomutagenic risk associated with photoreactive drugs. Moreover, comprehensive DNA damage analysis at different wavelengths is also required to identify the endogenous photosensitizers that are responsible for the DNA damage induced by UVA and visible light in mammalian cells and to estimate their contribution to the adverse effects of solar radiation.References
- S. Brendler-Schwaab, A. Czich, B. Epe, E. Gocke, B. Kaina, L. Muller, D. Pollet and D. Utesch, Photochemical genotoxicity: principles and test methods. Report of a GUM task force, Mutat. Res., 2004, 566, 65–91 CAS.
- J. Cadet, T. Douki and J. L. Ravanat, Measurement of oxidatively generated base damage in cellular DNA, Mutat. Res., 2011, 711, 3–12 CrossRef CAS.
- J. Cadet, E. Sage and T. Douki, Ultraviolet radiation-mediated damage to cellular DNA, Mutat. Res., 2005, 571, 3–17 CrossRef CAS.
- ESCODD, Measurement of DNA oxidation in human cells by chromatographic and enzymic methods, Free Radical Biol. Med., 2003, 34, 1089–1099 CrossRef.
- W. L. Neeley and J. M. Essigmann, Mechanisms of formation, genotoxicity, and mutation of guanine oxidation products, Chem. Res. Toxicol., 2006, 19, 491–505 CrossRef CAS.
- J. Musarrat and A. A. Wani, Quantitative immunoanalysis of promutagenic 8-hydroxy-2′-deoxyguanosine in oxidized DNA, Carcinogenesis, 1994, 15, 2037–2043 CrossRef CAS.
- Y. Nakae, P. J. Stoward, I. A. Bespalov, R. J. Melamede and S. S. Wallace, A new technique for the quantitative assessment of 8-oxoguanine in nuclear DNA as a marker of oxidative stress. Application to dystrophin-deficient DMD skeletal muscles, Histochem. Cell Biol., 2005, 124, 335–345 CrossRef CAS.
- B. Epe and J. Hegler, Oxidative DNA damage: endonuclease fingerprinting, Methods Enzymol., 1994, 234, 122–131 CAS.
- A. Hartwig, H. Dally and R. Schlepegrell, Sensitive analysis of oxidative DNA damage in mammalian cells: use of the bacterial Fpg protein in combination with alkaline unwinding, Toxicol. Lett., 1996, 88, 85–90 CrossRef CAS.
- A. Azqueta, K. B. Gutzkow, G. Brunborg and A. R. Collins, Towards a more reliable comet assay: Optimising agarose concentration, unwinding time and electrophoresis conditions, Mutat. Res., 2011, 724, 41–45 CAS.
- A. R. Collins, The use of bacterial repair endonucleases in the comet assay, Methods Mol. Biol., 2011, 691, 137–147 CAS.
- D. Averbeck, M. Dardalhon and N. Magana-Schwencke, Repair of furocoumarin-plus-UVA-induced damage and mutagenic consequences in eukaryotic cells, J. Photochem. Photobiol., B, 1990, 6, 221–236 CrossRef CAS.
- G. E. Pfyffer, B. U. Pfyffer and G. H. Towers, Monoaddition of dictamnine to synthetic double-stranded polydeoxyribonucleotides in UVA and the effect of photomodified DNA on template activity, Photochem. Photobiol., 1982, 35, 793–797 CrossRef CAS.
- E. Sage, T. Le Doan, V. Boyer, D. E. Helland, L. Kittler, C. Helene and E. Moustacchi, Oxidative DNA damage photo-induced by 3-carbethoxypsoralen and other furocoumarins. Mechanisms of photo-oxidation and recognition by repair enzymes, J. Mol. Biol., 1989, 209, 297–314 CrossRef CAS.
- F. Bosca, V. Lhiaubet-Vallet, M. C. Cuquerella, J. V. Castell and M. A. Miranda, The triplet energy of thymine in DNA, J. Am. Chem. Soc., 2006, 128, 6318–6319 CrossRef CAS.
- A. Moysan, A. Viari, P. Vigny, L. Voituriez, J. Cadet, E. Moustacchi and E. Sage, Formation of cyclobutane thymine dimers photosensitized by pyridopsoralens: quantitative and qualitative distribution within DNA, Biochemistry, 1991, 30, 7080–7088 CrossRef CAS.
- V. Lhiaubet-Vallet, M. C. Cuquerella, J. V. Castell, F. Bosca and M. A. Miranda, Triplet excited fluoroquinolones as mediators for thymine cyclobutane dimer formation in DNA, J. Phys. Chem. B, 2007, 111, 7409–7414 CrossRef CAS.
- K. S. Robinson, N. J. Traynor, H. Moseley, J. Ferguson and J. A. Woods, Cyclobutane pyrimidine dimers are photosensitised by carprofen plus UVA in human HaCaT cells, Toxicol. In Vitro, 2010, 24, 1126–1132 CrossRef CAS.
- D. Roca-Sanjuan, G. Olaso-Gonzalez, I. Gonzalez-Ramirez, L. Serrano-Andres and M. Merchan, Molecular basis of DNA photodimerization: intrinsic production of cyclobutane cytosine dimers, J. Am. Chem. Soc., 2008, 130, 10768–10779 CrossRef CAS.
- S. Mouret, C. Philippe, J. Gracia-Chantegrel, A. Banyasz, S. Karpati, D. Markovitsi and T. Douki, UVA-induced cyclobutane pyrimidine dimers in DNA: a direct photochemical mechanism?, Org. Biomol. Chem., 2010, 8, 1706–1711 CAS.
- D. Markovitsi, T. Gustavsson and A. Banyasz, Absorption of UV radiation by DNA: spatial and temporal features, Mutat. Res., 2010, 704, 21–28 CAS.
- B. Epe, H. Henzl, W. Adam and C. R. Saha-Moller, Endonuclease-sensitive DNA modifications induced by acetone and acetophenone as photosensitizers, Nucleic Acids Res., 1993, 21, 863–869 CrossRef CAS.
- W. Mu, Q. Han, Z. Luo and Y. Wang, Production of cis-syn thymine-thymine cyclobutane dimer oligonucleotide in the presence of acetone photosensitizer, Anal. Biochem., 2006, 353, 117–123 CrossRef CAS.
- S. Steenken and S. V. Jovanovic, How easily oxidizable is DNA? One-electron reduction potentials of adenosine and guanosine radicals in aqueous solution, J. Am. Chem. Soc., 1997, 119, 617–618 CrossRef CAS.
- K. Ito, S. Inoue, K. Yamamoto and S. Kawanishi, 8-Hydroxydeoxyguanosine formation at the 5′ site of 5′-GG-3′ sequences in double-stranded DNA by UV radiation with riboflavin, J. Biol. Chem., 1993, 268, 13221–13227 CAS.
- I. Saito, M. Takayama, H. Sugiyama and K. Nakatani, Photoinduced DNA Cleavage Via Electron-Transfer - Demonstration That Guanine Residues Located 5′ to Guanine Are the Most Electron-Donating Sites, J. Am. Chem. Soc., 1995, 117, 6406–6407 CrossRef CAS.
- H. Sugiyama and I. Saito, Theoretical studies of GC-specific photocleavage of DNA via electron transfer: Significant lowering of ionization potential and 5′-localization of HOMO of stacked GG bases in B-form DNA, J. Am. Chem. Soc., 1996, 118, 7063–7068 CrossRef CAS.
- J. Cadet, T. Douki and J. L. Ravanat, Oxidatively generated damage to the guanine moiety of DNA: mechanistic aspects and formation in cells, Acc. Chem. Res., 2008, 41, 1075–1083 CrossRef CAS.
- M. L. Wood, M. Dizdaroglu, E. Gajewski and J. M. Essigmann, Mechanistic studies of ionizing radiation and oxidative mutagenesis: genetic effects of a single 8-hydroxyguanine (7-hydro-8-oxoguanine) residue inserted at a unique site in a viral genome, Biochemistry, 1990, 29, 7024–7032 CrossRef CAS.
- K. C. Cheng, D. S. Cahill, H. Kasai, S. Nishimura and L. A. Loeb, 8-Hydroxyguanine, an abundant form of oxidative DNA damage, causes G—T and A—C substitutions, J. Biol. Chem., 1992, 267, 166–172 CAS.
- M. Moriya, Single-stranded shuttle phagemid for mutagenesis studies in mammalian cells: 8-oxoguanine in DNA induces targeted G.C–>T.A transversions in simian kidney cells, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 1122–1126 CrossRef CAS.
- D. Ballmaier and B. Epe, Oxidative DNA damage induced by potassium bromate under cell-free conditions and in mammalian cells, Carcinogenesis, 1995, 16, 335–342 CrossRef CAS.
- B. Matter, D. Malejka-Giganti, A. S. Csallany and N. Tretyakova, Quantitative analysis of the oxidative DNA lesion, 2,2-diamino-4-(2-deoxy-beta-D-erythro-pentofuranosyl)amino]-5(2H)-oxazolon e (oxazolone), in vitro and in vivo by isotope dilution-capillary HPLC-ESI-MS/MS, Nucleic Acids Res., 2006, 34, 5449–5460 CrossRef CAS.
- M. Dizdaroglu, G. Kirkali and P. Jaruga, Formamidopyrimidines in DNA: mechanisms of formation, repair, and biological effects, Free Radical Biol. Med., 2008, 45, 1610–1621 CrossRef CAS.
- I. Schulz, H. C. Mahler, S. Boiteux and B. Epe, Oxidative DNA base damage induced by singlet oxygen and photosensitization: recognition by repair endonucleases and mutagenicity, Mutat. Res., 2000, 461, 145–156 CAS.
- K. Kino, I. Saito and H. Sugiyama, Product analysis of GG-specific photooxidation of DNA via electron transfer: 2-aminoimidazolone as a major guanine oxidation product, J. Am. Chem. Soc., 1998, 120, 7373–7374 CrossRef CAS.
- K. Kino and H. Sugiyama, Possible cause of G–C–>C–G transversion mutation by guanine oxidation product, imidazolone, Chem. Biol., 2001, 8, 369–378 CrossRef CAS.
- M. Hariharan, S. C. Karunakaran, D. Ramaiah, I. Schulz and B. Epe, Photoinduced DNA damage efficiency and cytotoxicity of novel viologen linked pyrene conjugates, Chem. Commun., 2010, 46, 2064–2066 RSC.
- M. M. Gonzalez, M. Pellon-Maison, M. A. Ales-Gandolfo, M. R. Gonzalez-Baro, R. Erra-Balsells and F. M. Cabrerizo, Photosensitized cleavage of plasmidic DNA by norharmane, a naturally occurring beta-carboline, Org. Biomol. Chem., 2010, 8, 2543–2552 CAS.
- C. S. Foote, Definition of type I and type II photosensitized oxidation, Photochem. Photobiol., 1991, 54, 659 CrossRef CAS.
- F. Bergeron, F. Auvre, J. P. Radicella and J. L. Ravanat, HO* radicals induce an unexpected high proportion of tandem base lesions refractory to repair by DNA glycosylases, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 5528–5533 CrossRef CAS.
- J. Cadet, T. Douki, D. Gasparutto and J. L. Ravanat, Oxidative damage to DNA: formation, measurement and biochemical features, Mutat. Res., 2003, 531, 5–23 CrossRef CAS.
- J. P. Pouget, S. Frelon, J. L. Ravanat, I. Testard, F. Odin and J. Cadet, Formation of modified DNA bases in cells exposed either to gamma radiation or to high-LET particles, Radiat. Res., 2002, 157, 589–595 CrossRef CAS.
- K. W. Kohn, L. C. Erickson, R. A. Ewig and C. A. Friedman, Fractionation of DNA from mammalian cells by alkaline elution, Biochemistry, 1976, 15, 4629–4637 CrossRef CAS.
- M. Häring, H. Rudiger, B. Demple, S. Boiteux and B. Epe, Recognition of oxidized abasic sites by repair endonucleases, Nucleic Acids Res., 1994, 22, 2010–2015 CrossRef.
- B. Epe, D. Ballmaier, W. Adam, G. N. Grimm and C. R. Saha-Moller, Photolysis of N-hydroxpyridinethiones: a new source of hydroxyl radicals for the direct damage of cell-free and cellular DNA, Nucleic Acids Res., 1996, 24, 1625–1631 CrossRef CAS.
- B. Epe, M. Haring, D. Ramaiah, H. Stopper, M. M. Abou-Elzahab, W. Adam and C. R. Saha-Moller, DNA damage induced by furocoumarin hydroperoxides plus UV (360 nm), Carcinogenesis, 1993, 14, 2271–2276 CrossRef CAS.
- H. C. Mahler, I. Schulz, W. Adam, G. N. Grimm, C. R. Saha-Moller and B. Epe, tert-Butoxyl radicals generate mainly 7,8-dihydro-8-oxoguanine in DNA, Mutat. Res., 2001, 461, 289–299 CAS.
- Y. Ye, J. G. Muller, W. Luo, C. L Mayne, A. J. Shallop, R. A. Jones and C. J. Burrows, Formation of 13C-, 15N-, and 18O-labeled guanidinohydantoin from guanosine oxidation with singlet oxygen. Implications for structure and mechanism, J. Am. Chem. Soc., 2003, 125, 13926–13927 CrossRef CAS.
- J. E. McCallum, C. Y. Kuniyoshi and C. S. Foote, Characterization of 5-hydroxy-8-oxo-7,8-dihydroguanosine in the photosensitized oxidation of 8-oxo-7,8-dihydroguanosine and its rearrangement to spiroiminodihydantoin, J. Am. Chem. Soc., 2004, 126, 16777–16782 CrossRef CAS.
- J. L. Ravanat, P. Di Mascio, G. R. Martinez, M. H. Medeiros and J. Cadet, Singlet oxygen induces oxidation of cellular DNA, J. Biol. Chem., 2000, 275, 40601–40604 CrossRef CAS.
- J. L. Ravanat, S. Sauvaigo, S. Caillat, G. R. Martinez, M. H. Medeiros, P. Di Mascio, A. Favier and J. Cadet, Singlet oxygen-mediated damage to cellular DNA determined by the comet assay associated with DNA repair enzymes, Biol. Chem., 2004, 385, 17–20 CrossRef CAS.
- E. Muller, S. Boiteux, R. P. Cunningham and B. Epe, Enzymatic recognition of DNA modifications induced by singlet oxygen and photosensitizers, Nucleic Acids Res., 1990, 18, 5969–5973 CrossRef CAS.
- O. Will, E. Gocke, I. Eckert, I. Schulz, M. Pflaum, H. C. Mahler and B. Epe, Oxidative DNA damage and mutations induced by a polar photosensitizer, Ro19-8022, Mutat Res., 1999, 435, 89–101 CAS.
- M. E. Serrentino, A. Catalfo, A. R. Angelin, G. de Guidi and E. Sage, Photosensitization induced by the antibacterial fluoroquinolone Rufloxacin leads to mutagenesis in yeast, Mutat Res., 2010, 692, 34–41 CrossRef CAS.
- E. Gocke, Review of the genotoxic properties of chlorpromazine and related phenothiazines, Mutat Res., 1996, 366, 9–21 Search PubMed.
- T. A. Ciulla, G. A. Epling and I. E. Kochevar, Photoaddition of chlorpromazine to guanosine-5′-monophosphate, Photochem. Photobiol., 1986, 43, 607–613 CrossRef CAS.
- A. W. Girotti, Photosensitized oxidation of membrane lipids: reaction pathways, cytotoxic effects, and cytoprotective mechanisms, J. Photochem. Photobiol., B, 2001, 63, 103–113 CrossRef CAS.
- L. J. Marnett, Oxyradicals and DNA damage, Carcinogenesis, 2000, 21, 361–370 CrossRef CAS.
- H. Bartsch and J. Nair, Ultrasensitive and specific detection methods for exocylic DNA adducts: markers for lipid peroxidation and oxidative stress, Toxicology, 2000, 153, 105–114 CrossRef CAS.
- I. G. Minko, I. D. Kozekov, T. M. Harris, C. J. Rizzo, R. S. Lloyd and M. P. Stone, Chemistry and biology of DNA containing 1,N(2)-deoxyguanosine adducts of the alpha,beta-unsaturated aldehydes acrolein, crotonaldehyde, and 4-hydroxynonenal, Chem. Res. Toxicol., 2009, 22, 759–778 CrossRef CAS.
- G. P. Voulgaridou, I. Anestopoulos, R. Franco, M. I. Panayiotidis and A. Pappa, DNA damage induced by endogenous aldehydes: Current state of knowledge, Mutat. Res., 2011, 711, 13–27 CrossRef CAS.
- C. Kielbassa, L. Roza and B. Epe, Wavelength dependence of oxidative DNA damage induced by UV and visible light, Carcinogenesis, 1997, 18, 811–816 CrossRef CAS.
- J. P. Pouget, T. Douki, M. J. Richard and J. Cadet, DNA damage induced in cells by gamma and UVA radiation as measured by HPLC/GC-MS and HPLC-EC and Comet assay, Chem. Res. Toxicol., 2000, 13, 541–549 CrossRef CAS.
- T. Douki, A. Reynaud-Angelin, J. Cadet and E. Sage, Bipyrimidine photoproducts rather than oxidative lesions are the main type of DNA damage involved in the genotoxic effect of solar UVA radiation, Biochemistry, 2003, 42, 9221–9226 CrossRef CAS.
- S. Courdavault, C. Baudouin, M. Charveron, A. Favier, J. Cadet and T. Douki, Larger yield of cyclobutane dimers than 8-oxo-7,8-dihydroguanine in the DNA of UVA-irradiated human skin cells, Mutat. Res., 2004, 556, 135–142 CrossRef CAS.
- M. Pflaum, C. Kielbassa, M. Garmyn and B. Epe, Oxidative DNA damage induced by visible light in mammalian cells: extent, inhibition by antioxidants and genotoxic effects, Mutat. Res., 1998, 408, 137–146 CAS.
- G. T. Wondrak, M. K. Jacobson and E. L. Jacobson, Endogenous UVA-photosensitizers: mediators of skin photodamage and novel targets for skin photoprotection, Photochem. Photobiol. Sci., 2006, 5, 215–237 CAS.
- B. Epe, Role of endogenous oxidative DNA damage in carcinogenesis: what can we learn from repair-deficient mice?, Biol. Chem., 2002, 383, 467–475 CrossRef CAS.
- S. Hoffmann-Dörr, R. Greinert, B. Volkmer and B. Epe, Visible light (>395 nm) causes micronuclei formation in mammalian cells without generation of cyclobutane pyrimidine dimers, Mutat. Res., 2005, 572, 142–149 CrossRef.
- M. A. Bachelor and G. T. Bowden, UVA-mediated activation of signaling pathways involved in skin tumor promotion and progression, Semin. Cancer Biol., 2004, 14, 131–138 CrossRef CAS.
- S. Boiteux and M. Guillet, Abasic sites in DNA: repair and biological consequences in Saccharomyces cerevisiae, DNA Repair, 2004, 3, 1–12 CrossRef CAS.
- S. Bjelland and E. Seeberg, Mutagenicity, toxicity and repair of DNA base damage induced by oxidation, Mutat. Res., 2003, 531, 37–80 CrossRef CAS.
- H. E. Krokan, R. Standal and G. Slupphaug, DNA glycosylases in the base excision repair of DNA, Biochem. J., 1997, 325(Pt 1), 1–16 CAS.
- M. Dizdaroglu, Base-excision repair of oxidative DNA damage by DNA glycosylases, Mutat. Res., 2005, 591, 45–59 CrossRef CAS.
- W. Eiberger, B. Volkmer, R. Amouroux, C. Dherin, J. P. Radicella and B. Epe, Oxidative stress impairs the repair of oxidative DNA base modifications in human skin fibroblasts and melanoma cells, DNA Repair, 2008, 7, 912–921 CrossRef CAS.
Footnote |
| † Contribution to the themed issue on the biology of UVA. |
| This journal is © The Royal Society of Chemistry and Owner Societies 2012 |