Cationic and radical intermediates in the acid photorelease from aryl sulfonates and phosphates†
Marco
Terpolilli
a,
Daniele
Merli
b,
Stefano
Protti
a,
Valentina
Dichiarante
a,
Maurizio
Fagnoni
*a and
Angelo
Albini
a
aDepartment of Organic Chemistry, University of Pavia, Pavia, 27100, Italy. E-mail: fagnoni@unipv.it
bDepartment of General Chemistry, University of Pavia, Pavia, 27100, Italy
First published on 9th November 2010
Abstract
The irradiation of a series of phenyl sulfonates and phosphates leads to the quantitative release of acidity with a reasonable quantum yield (≈0.2). Products characterization, ion chromatography analysis and potentiometric titration are consistent with the intervening of two different paths in this reaction, viz. cationic with phosphates and (mainly) radical with sulfonates.
Introduction
Molecules capable of generating acidity upon irradiation (Photo Acid Generators, PAGs), are used as initiators in cationic polymerization, in chemically amplified photoresists,1a–c as well as in bioorganic synthesis.1d–e The most widely used class of these compounds is that of aromatic onium salts ArnX+MYm−, such as iodonium and sulfonium salts.2 These are a convenient choice because of their thermal stability (>200 °C) and the easy chemical modification that allows tuning the absorption characteristics. Some drawbacks, such as the limited solubility in organic solvents that may cause phase separation from spun-on resist polymer matrices, make the development of new “caged protons” desirable, with particular regard to non-ionic molecules. The most important example of the latter class are the diazonaphthoquinone derivatives (DNQ), where a carboxylic acid is liberated after a photo-Wolff rearrangement.1a The photorelease of other acids (e.g. sulfonic) has been more elusive and involves substituted esters. Mechanisms that have been considered include the photoinduced intramolecular hydrogen abstraction in 2-nitrobenzyl sulfonates3 and the homolytic photocleavage of the ArO–S bond in aryl sulfonates. In the latter case, the initial step leads either to (non-acid-generating) photo-Fries rearrangement or to hydrogen abstraction from the solvent, depending on the structure.4 Polysulfonates, e.g. the tris(mesylate) of pyrogallol (MeSB), release more acidity than monosulfonates,5 and indeed MeSB-based resists were found to be more photosensitive than those incorporating a sulfonium salt.6 In recent years, our own and other groups have been studying the photochemistry of electron-rich aromatic halides and esters (1 in Scheme 1) in polar solvents and found that heterolysis of the Ar–X bond occurs, most effectively under acetone sensitization, and leads to triplet phenyl cations.7–9 These are synthetically useful intermediates that have been exploited for a variety of arylation reactions. | ||
| Scheme 1 Photo acid generation viaphenyl cations. | ||
Organic preparations by this method are usually carried out under buffered conditions, because otherwise the solution pH strongly decreases (Scheme 1). The latter characteristics is appealing because, if the reaction occurs according to Scheme 1, it should be possible not only to generate acidity from such compounds, but also to tune the strength of the photogenerated acid by changing the group X in the precursor.
As an example, triflic (pKa = −13), hydrochloric (pKa = −6), methanesulfonic (pKa = −1.54), diethylphosphoric (pKa = 0.71) and hydrofluoric acid (pKa = 3.18) could be photoreleased as preferred from compounds of formula 1.
In the following, a study aimed to explore the potentialities of this class of compounds for the photorelease of acids is reported. Some families of aromatic derivatives were examined, of which the amount and nature of the acid liberated had not been previously determined. In the choice of the potential PAGs to explore this issue, those liberating volatile acids such as HF and HCl that can cause pinholes in the resist films were excluded. Sulfonates were thus preferred, on the basis of the potential release of strong acids, including triflic acid, a somewhat volatile but strong acid10 that is known for inducing acid amplification in photoresists.11 It was appealing also to investigate phosphate esters, at they are presently scarcely considered.12,13
Materials and methods
Previous studies have shown that phenyl cations are conveniently photogenerated starting from various electron-donating substituted aromatics.10 However, aminophenol and hydroxyphenol esters were not considered for this work due to their poor thermal stability and easy oxidation. Thus, sulfonate and phosphate esters of p-methoxy (1a,c) and p-t-butylphenol (1d,f) were chosen.Experimental details: general
Unless otherwise stated, all reagents were commercially available and used without purification. 1H and 13C NMR spectra were recorded on a 300 MHz (Bruker) spectrometer. The attributions were made on the basis of 1H and 13C NMR; chemical shifts are reported in ppm downfield from TMS. GC-MS analyses have been carried out with a Thermo Scientific DSQ II Single Quadrupole instrument (Column Rtx-5 MS (Restek), Detection mode: Electronic Impact).Synthesis of esters 1a–f
p-Methoxy esters 1a–c were obtained as previously described.8Mesylate 1d, triflate 1e and phosphate 1f were obtained by known procedures from the corresponding phenols. 4-tert-Butylphenylmethanesulfonate (1d) was obtained from the corresponding phenol by a known procedure14 in 56% yield as a colorless solid, mp 49–50 °C (from ethanol, lit: 50–52 °C15). Spectroscopic data of 1d in accordance with literature data.15 Anal. calcd for C11H16O3S: C, 57.87; H, 7.06%. Found: C, 57.9; H, 7.1%. 4-tert-Butylphenyltrifluoromethanesulfonate (1e) was synthesized from p-(t-butyl)phenol in 43% yield as an oil.16 Spectroscopic data of 1e in accordance with literature data.17 Anal. calcd for C11H13F3O3S: C, 46.80; H, 4.64%. Found: C, 46.9; H, 4.6%. 4-tert-Butylphenyldiethylphosphate (1f) was obtained by a known procedure18 in 69% yield as an oil. 1H NMR (δ, CDCl3):19 1.30 (s, 9H), 1.35–1.40 (t, 6H, J = 6 Hz), 4.20–4.30 (qui, J = 6 Hz), 7.10–7.35 (AA′BB′, 4H). 13C NMR (δ, CDCl3): 15.9 (CH3), 31.3 (CH3), 34.2, 64.3 (CH2), 119.2 (CH), 126.4 (CH), 147.7, 148.3. IR (neat, ν/cm−1: 2965, 1513, 1272, 1031, 964, 840. Anal. calcd for C14H13O4P: C, 58.73; H, 8.10%. Found: C, 58.7; H, 8.0%.Photolysis of esters 1a–f
The photochemical reactions were performed by using nitrogen-purged solutions (except where indicated) of the chosen ester (0.01 M) in quartz tubes. HPLC grade purity methanol (0.05% water content as determined by Karl-Fischer titration) was used as solvent for all the photolysis experiments. Irradiations were performed in a multilamp reactor fitted with 4 low pressure Hg lamps (emission maximum: λ = 254 nm), and the reaction course was followed by GC analysis. As described below, organic photoproducts from 1a,f have been quantified on the basis of calibration curves from commercial samples. The presence of gaseous fluoroform in the photolyzed solution has been revealed by means of GC-MS and IR analysis (gas, ν/cm−1 3035, 1374, 115820). In the last case, the analysis was performed on the gas released in a 10 cm cell by using sodium chloride windows, internal volume 100 ml, purged with nitrogen before the introduction of the sample.The acidity liberated has been determined on 10 mL of the photolysed solutions by dilution with water (50 mL) and titration with aqueous 0.1 M NaOH. Potentiometric acid titrations (NaOH 0.1 M) of 1b–e have been followed by using an Orion mod. 250 potentiometer equipped with an Orion pH glass combined electrode mod 91–56. The titrations by using 0.1 M NaOH have been performed on 10 mL photolyzed solutions in MeOH diluted with 100 mL water.
Ion chromatography analyses have been performed by means of a Dionex GP40 instrument equipped with a conductimetric detector (Dionex 20 CD20) and an electrochemical suppressor (ASRS Ultra II, 4 mm) by using the following conditions: chromatographic column IONPAC AS23 (4 mm × 250 mm), guard column IONPAC AG12 (4 mm × 50 mm), eluent: NaHCO3 0.8 mM + Na2CO3 4.5 mM, flux: 1 mL min−1; current imposed at detector: 50 mA. Commercially available tetrabutylammonium mesylate, trifluoromethanesulfonic acid and diethyl phosphate were used as standards.
Results and discussion
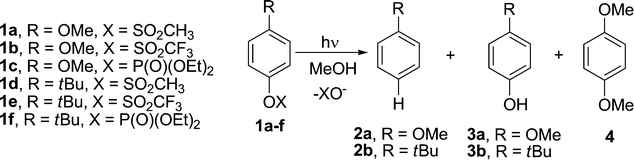
Photophysical and photochemical properties of esters 1a,f have been resumed in Table 1. Methoxyphenyl esters 1a–c exhibited two absorption maxima in the UV around 276 (ε > 103 L cm−1 mol−1) and 224 nm (ε ≈ 104 L cm−1 mol−1), while t-butylphenyl esters 1d–f absorbed somewhat blue-shifted and less intensively (260, ε > 102 L cm−1 mol−1, and 212 nm, ε ≈ 104 L cm−1 mol−1 as the maxima, see Table 1 and Fig. 1).|
|
||||||
|---|---|---|---|---|---|---|
| Ester | λmax/nm (ε/L cm−1 mol−1) | tir/ha | ΦR | Photoproducts, yield (%)b | H+ yield (%)c | Anions, yield (%)c,d |
| a 0.01 M solution of 1a–f in degassed methanol, irradiated at 254 nm until complete consumption of the ester. b GC yield. c The analysis is based on a 0.03 mmol amount of starting 1. d Variable amounts of formate anion have been detected in all the photolyzed solutions. e Fluoroform was detected by IR analysis. f Reaction carried out in oxygen saturated solution. g See ref. 14. h Fluoroform was detected by GC-MS analysis. | ||||||
| 1a | 276 (1210) | 6 | 0.13 | 2a, 19; 3a, 25; 4, 9 | 95.0 | CH3SO3−, 46.7 |
| 224 (7530) | ||||||
| 1b | 275 (1855) | 6 | 0.2 | 3a, 51 | 95.0 | CF3SO3−, 1.2e (31.7)f; |
| 225 (12170) | F−, 36.7 (8.0)f | |||||
| 1c | 279 (2395) | 6 | 0.16g | 2a, 58; 4, 25 | 98.3 | (EtO)2PO2−, 100 |
| 224 (8934) | ||||||
| 1d | 261 (240) | 6 | 0.17 | 3b, 68 | 95.0 | CH3SO3−, 93.3 |
| 214 (8112) | ||||||
| 1e | 258 (214) | 6 | 0.11 | 2b, 5; 3b, 60 | 93.3 | CF3SO3−, 0h (56.7)f; |
| 212 (9643) | F−, 56.7 (25.7)f | |||||
| 1f | 265 (530) | 24 | 0.02 | 2b, 36 | 90.0 | (EtO)2PO2−, 93.3 |
| 211 (7750) | ||||||
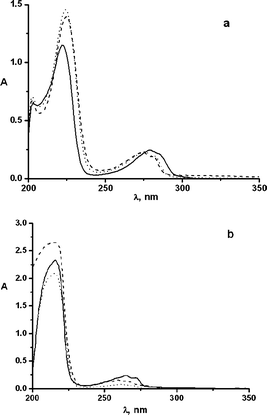 | ||
| Fig. 1 Absorption spectra of (a) 1 × 10−4 M MeOH solution of 1a (dotted line), 1b (dashed line), 1c (solid line) and (b) 5 × 10−4 M MeOH solution of 1d (dotted line), 1e (dashed line) and 1f (solid line). | ||
In this study, 0.01 M methanol solutions of esters 1a–f were de-aerated and irradiated, resulting in complete consumption after 6 h in almost all cases. The quantum yield of disappearance of 1 (ΦR), at 254 nm ranged from 0.11 to 0.2, except for the case of less reactive phosphate 1f (ΦR = 0.02).
The percent yield of organic products and the mmol of inorganic products formed upon 6 h irradiation (24 h for 1f) of solutions containing 0.3 mmol of starting compound 1, as well as the acidity generated are reported in Table 1.
The pH of the photolyzed solutions was around 1 and we were happy to find that the amount of acid released was close to quantitative (>90%) as titrated by NaOH.
More precisely, potentiometric titrations evidenced a single pKa value for 1c and 1d, but two for 1b and 1e (ratio between the stronger and the weaker acid 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 2:1 respectively, see Fig. 2).
:1 and 2:1 respectively, see Fig. 2).
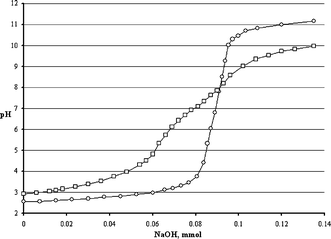 | ||
| Fig. 2 Potentiometric titration of a 10−2 M solution of 1c (○, 0.1 mmol) and 1e (□, 0.1 mmol) after irradiation in MeOH. | ||
As for the organic moiety, the photoprocesses occurring are reduction (giving 2), formal hydrolysis (forming deprotected 3) and solvolysis (4), since no photo-Fries adducts were detected by GC analysis. Triflates 1b, 1e gave the corresponding phenol 3 as the exclusive (from 1b) or the main photoproduct (1e gave also a 5% amount of t-butylbenzene 2b). In the case of p-methoxyphenyl mesylate (1a), deprotection and reduction took place in a comparable yield (25% vs. 19%), but some dimethoxybenzene was formed too (4, 9%), while the other mesylate (1e) was exclusively deprotected to give p-t-butylphenol 3b.
With phosphates 1c and 1f photoreduction was the main process giving anisole 2a (58%, along with ether 4 in 25% yield) and alkylbenzene 2b (36%), respectively.
According to ion chromatography analyses, phosphates and mesylates (except 1a) released the O,O-diethylphosphate and the mesylate anion, respectively in a close to quantitative yield (>92%). On the contrary, only traces of triflate anion were present in photolyzed solutions of 1b,1e, where the main anion was fluoride (up to 56% yield). In this case, a gas was liberated, as apparent from the fizz on opening the reaction tubes, that was shown to be fluoroform, CHF3 by IR20 and GC-MS analysis.
It is noteworthy that, when the photolysis of 1b,1e was performed in oxygen saturated methanol, triflate anion was formed in a good yield at the expense of fluoride (Table 1).
In the case of 1e, small amounts of sulfur dioxide, sulfurous acid dimethyl ester as well as dimethoxymethane, ethandiol and 4-t-butylanisole were also detected. Moreover, variable amounts of formate anion have been detected in all of the irradiated solutions.
The data above demonstrate that all of the aryl esters release acidity with a reasonable quantum yields (except for 1f). The thermal stability, good miscibility with resins and convenient (and tunable) absorption in the UV-C make these compounds suitable candidates as PAGs. It is noteworthy that two different mechanisms operate involving a cationic and a radical intermediates, as shown in Schemes 2 and 3. With phosphates 1c,f, intersystem crossing (ISC) is efficient,21 and product analysis supports that the reaction proceeds from the triplet state and involves heterolysis of the aryl-oxygen bond (cleavage a, Scheme 2). Triplet phenyl cation (31+) and the corresponding counterion (EtO)2PO2− are formed. As previously demonstrated,22 the cation abstracts a hydrogen atom from the medium and gives anisole (2a) or t-butylbenzene (2b) via radical cation 5 along with formaldehyde that under acidic conditions is transformed into dimethoxymethane or (photo)oxidized23 to formic acid (Scheme 2).
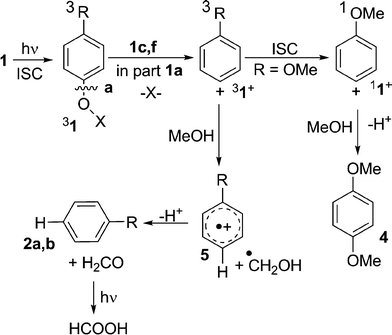 | ||
| Scheme 2 Cationic intermediates in the photolysis of 1. | ||
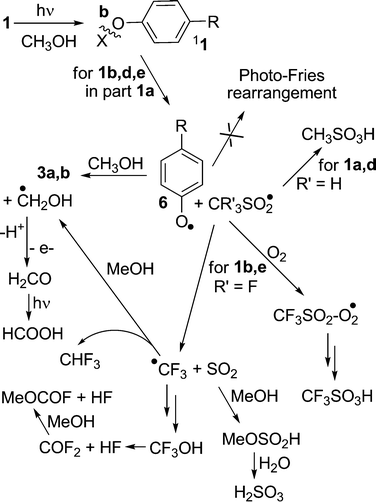 | ||
| Scheme 3 Radical intermediates in the photolysis of 1. | ||
In part, the triplet cation intersystem crosses to the singlet (11+, R = OMe)9 that in turn adds the solvent and gives p-dimethoxybenzene (4).24 The last pathway scarcely contributes in the case of triplet 4-t-butylphenyl cation, which rather abstracts hydrogen and thus gives 2b with only a trace of the solvolysis product, 4-t-butylanisole.23 The heterolytic path accounts also for part of the reaction of mesylate 1a, as witnessed by the formation of products 2a and 4.
In the aim of confirming the proposed mechanism, irradiations experiments in deuterated solvents have been carried out. Thus, photolysis of phosphate 1c in CH3OD afforded non-deuterated anisole 2a exclusively. On the contrary, in CD3OD complete monodeuterated anisole 2a-d1, was formed along with trideuterated 4-d3.‡ On the other hand, among sulfonates homolytic cleavage of the oxygen-sulfur bond competes (in the case of 1a) or becomes the exclusive path (with mesylate 1d and triflates 1b,e). Since heterolytic cleavage is the exclusive reaction in the acetone sensitized irradiation of 1a and 1b,8 homolytic cleavage proceeds from the singlet. A phenoxyl (6) and a sulfonyl radical are formed (cleavage b, Scheme 3)25 and escape from the solvent cage (no photo-Fries products are detected). Radical 6 abstracts hydrogen and gives the corresponding phenol 3 along with a ˙CH2OH radical that ultimately is converted into formic acid or dimerized to form ethandiol. As for the sulfonyl radical CR′3SO2˙, its fate depends on the nature of group CR′3. The CH3SO2˙ radical gives methanesulfonic acid,26 while in the case of trifluoromethanesulfonyl radical such a path is overcome by the known α cleavage to trifluoromethyl radical CF3˙ and SO2.27,28 The sulfur dioxide formed is either liberated as a gas or reacts with the solvent giving MeOSO2H29 (and in part the dimethyl ester30). This accounts for the formation of a certain amount of weak sulfurous acid, as evidenced in the potentiometric titration. The acid is formed upon dilution of the photolyzed solution with water (see ESI†). As for the CF3˙ radical, this abstracts hydrogen from the solvent giving inert fluoroform as the final product31 or CF3OH.31,32 The last compound is known to fragment thermally to HF and COF233 that liberates a further amount of HF by reaction with MeOH.34
The case with oxygenated solutions is different, where the radical CF3SO2˙ is trapped by O235 and gives triflic acidvia the peroxidic intermediate CF3SO2–O2˙.
Conclusions
The promising characteristics of a number of aryl esters for acid photorelease have been demonstrated. Besides the homolytic cleavage of the oxygen-sulfur bond, presumably operating also in some known PAGs as pyrogallol trimesylate, heterolytic cleavage of the aryl-oxygen bond has been demonstrated to contribute. The latter path had not been previously invoked among PAGs and its participation depends on the nature of the leaving group and of the aromatic moiety. Such a path can be made more important, at least with methoxyphenyl sulfonate esters, by passing to triplet sensitized conditions.8 In contrast, the heterolytic path is exclusive with phosphates and does not require sensitization. When the homolytic path is followed with triflates, the role of oxygen is determining for forming a strong acid and avoiding the release of volatile substances (HF and CHF3) that can have an adverse effect in a photoresist.Acknowledgements
We are grateful to Dr Barbara Mannucci and Dr Federica Corana of Centro Grandi Strumenti (University of Pavia) for technical support.Notes and references
- (a) S.-Y. Moon and J.-M. Kim, Chemistry of photolithographic imaging materials based on the chemical amplification concept, J. Photochem. Photobiol. C, 2007, 8, 157–173 CAS; (b) J. M. Frechét, The photogeneration of acid and base within polymer coatings: Approaches to polymer curing and imaging, Pure Appl. Chem., 1992, 64, 1239–1248 CrossRef CAS; (c) M. Shirai and M. Tsunooka, Photoacid and photobase generators: Chemistry and applications to polymeric materials, Prog. Polym. Sci., 1996, 21, 1–45 CrossRef CAS; (d) P. J. Serafinowski and P. B. Garland, Novel photoacid generators for photodirected oligonucleotide synthesis, J. Am. Chem. Soc., 2003, 125, 962–965 CrossRef CAS; (e) X. Gao, E. Gulari and X. Zhou, In situ synthesis of oligonucleotide microarrays, Biopolymers, 2004, 73, 579–596 CrossRef CAS.
- J. V. Crivello, The discovery and development of onium salt cationic photoinitiators, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 4241–4254 CrossRef CAS.
- F. M. Houlihan, O. Nalamasu, J. Kometani and E. Reichmanis, A retrospective on 2-nitrobenzyl sulfonate photoacid generators, J. Imag. Sci. Technol., 1997, 41, 35–40 Search PubMed.
- Y.-S. Chang, J.-S. Jang and M.-L. Deinzer, Photochemistry of irgasan-triflate: A simple conversion of an aromatic hydroxyl group to chlorine in the synthesis of polychlorinated diphenyl ethers and polychlorinated dibenzofurans, Tetrahedron, 1990, 46, 4161–4164 CrossRef CAS.
- (a) T. Ueno, L. Schlegel, N. Hayashi, H. Shiraishi and T. Iwayanagi, Acid formation from various sulfonates in a chemical amplification positive resist, Polym. Eng. Sci., 1992, 32, 1511–1515 CrossRef CAS; (b) L. Schlegel, T. Ueno, H. Shiraishi, N. Hayashi and T. Iwayanagi, Acid formation and deprotection reaction by novel sulfonates in a chemical amplification positive photoresist, Chem. Mater., 1990, 2, 299–305 CrossRef CAS.
- L. Schlegel, T. Ueno, H. Shiraishi, N. Hayashi and T. Iwayanagi, Highly sensitive positive deep UV resist utilizing a sulfonate acid generator and a tetrahydropyranyl inhibitor, Microelectron. Eng., 1991, 13, 33–36 CrossRef CAS.
- (a) V. Dichiarante and M. Fagnoni, Aryl cation chemistry as an emerging versatile tool for metal-free arylations, Synlett, 2008, 787–800 CAS; (b) M. Fagnoni and A. Albini, Arylation reactions: the photo- SN1 path via phenyl cation as an alternative to metal catalysis, Acc. Chem. Res., 2005, 38, 713–721 CrossRef CAS; (c) M. Slegt, S. Hermen, H. S. Overkleeft and G. Lodder, Fingerprints of singlet and triplet phenyl cations, Eur. J. Org. Chem., 2007, 5364–5375 CrossRef CAS.
- M. De Carolis, S. Protti, M. Fagnoni and A. Albini, Metal-free cross-coupling reactions of aryl sulfonates and phosphates through photoeterolysis of aryl-oxygen bonds, Angew. Chem., Int. Ed., 2005, 44, 1232–1236 CrossRef.
- S. Lazzaroni, D. Dondi, M. Fagnoni and A. Albini, Geometry and energy of substituted phenyl cations, J. Org. Chem., 2008, 73, 206–211 CrossRef CAS.
- R. D. Allen, J. Opitz, C. E. Larson, T. I. Wallow, R. A. DiPietro, G. Breyta, R. Sooriyakumaran and D. C. Hofer, The influence of photoacid structure on the design and performance of 193 nm resists, J. Photopolym. Sci. Technol., 1997, 10, 503–510 CrossRef CAS.
- (a) S. Lee, K. Arimitsu, S.-W. Park and K. Ichimura, Synthesis and evaluation of novel acid amplifiers liberating fluoroalkanesulfonic acids, J. Photopolym. Sci. Technol., 2000, 13, 215–216 CrossRef CAS; (b) S. Kruger, S. Revuru, C. Higgins, S. Gibbons, D. A. Freedman, W. Yueh, T. R. Younkin and R. L. Brainard, Fluorinated acid amplifiers for EUV lithography, J. Am. Chem. Soc., 2009, 131, 9862–9863 CrossRef CAS.
- S. Protti and M. Fagnoni, Phosphate esters as “tunable” reagents in organic synthesis, Chem. Commun., 2008, 3611–3621 RSC.
- M. Shi, K. Yamamoto, Y. Okamoto and S. Takamuku, Photolysis of aryl esters of tri- and tetracoordinate phosphorus compounds, Phosphorus, Sulfur Silicon Relat. Elem., 1991, 60, 1–14 CAS.
- R. K. Crossland and K. L. Servis, Facile synthesis of methanesulfonate esters, J. Org. Chem., 1970, 35, 3195–3196 CrossRef CAS.
- R. H. Munday, J. R. Martinelli and S. L. Buchwald, Palladium-catalyzed carbonylation of aryl tosylates and mesylates, J. Am. Chem. Soc., 2008, 130, 2754–2755 CrossRef CAS.
- D. E. Frantz, D. G. Weaver, J. P. Carey, M. H. Kress and U. H. Dolling, Practical synthesis of aryl triflates under aqueous conditions, Org. Lett., 2002, 4, 4717–4718 CrossRef CAS.
- (a) F. Y. Kwong, C. W. Lai, M. Yu, Y. Tian and K. S. Chan, Palladium-catalyzed phosphination of functionalized aryl triflates, Tetrahedron, 2003, 59, 10295–10305 CrossRef CAS; (b) W. M. Seganish and P. DeShong, Preparation and palladium-catalyzed cross-coupling of aryl triethylammonium bis(catechol) silicates with aryl triflates, J. Org. Chem., 2004, 69, 1137–1143 CrossRef CAS.
- K. Genkina, A. E. Shipov, T. A. Mastryukova and M. I. Kabachnik, Synthesis of unsymmetrical triaryl phosphates under phase-transfer catalysis, Russ. J. Gen. Chem., 1996, 66, 1742–1744.
- R. Tacke, M. Strecker and R. Niedner, Sila-Pharmaka, 22. Cholinesterase-hemmende organophosphorsäureester und ihre sila-analoga, Liebigs Ann. Chem., 1981, 1981, 387–395 CrossRef.
- R. A. Levine and W. B. Person, Absolute infrared intensity measurements of fluoroform (CHF3) fundamentals, J. Phys. Chem., 1977, 81, 1118–1119 CrossRef CAS.
- V. Dichiarante, D. Dondi, S. Protti, M. Fagnoni and A. Albini, A meta effect in organic photochemistry?The case of SN1 reactions in methoxyphenyl derivatives, J. Am. Chem. Soc., 2007, 129, 5605–5611 CrossRef CAS , correction: 11662.
- V. Dichiarante, M. Fagnoni and A. Albini, Eco-friendly hydrodehalogenation of electron-rich aryl chlorides and fluorides by photochemical reaction, Green Chem., 2009, 11, 942–945 RSC.
- O. A. Snytnikova, A. N. Simonov, O. P. Pestunova, V. N. Parnom and Y. P. Tsentalovich, Study of the photoinduced formose reaction by flash and stationary photolysis, Mendeleev Commun., 2006, 16, 9–11 Search PubMed.
- Studies on the photodecomposition of phenyldiazonium ions demonstrated that singet phenyl cations add non-selectively even to weak nucleophiles. See: S. Milanesi, M. Fagnoni and A. Albini, Cationic arylation through photo(sensitised) decomposition of diazonium salts. Chemoselectivity of triplet phenyl cations, Chem. Commun., 2003, 216–217 Search PubMed.
- See, for example: J. Andraos, G. G. Barclay, D. R. Medeiros, M. V. Baldovi, J. C. Scaiano and R. Sinta, Model studies on the photochemistry of phenolic sulfonate photoacid generators, Chem. Mater., 1998, 10, 1694–1699 Search PubMed.
- E. Robert-Banchereau, S. Lacombe and J. Ollivier, Unsensitized photooxidation of sulfur compounds with molecular oxygen in solution, Tetrahedron, 1997, 53, 2087–2102 CrossRef CAS.
- F. Bertrand, F. Leguyader, L. Liguori, G. Ouvry, B. Quiclet-Sire, S. Seguin and S. Z. Zard, α-Scission of sulfonyl radicals: a versatile process for organic synthesis, C. R. Acad. Sci. II C, 2001, 4, 547–555 Search PubMed.
- D. Gonbeau, M. F. Guimon, S. Duplantier, J. Ollivier and G. Pfister-Guillouzo, Sulfochloration of trifluoromethane. Theoretical study of the trifluoromethane sulfonyl radical, Chem. Phys., 1989, 135, 85–89 CrossRef CAS.
- L. S. Guss and I. M. Kolthoff, The behavior of sulfur dioxide as an acid in methanol, J. Am. Chem. Soc., 1944, 66, 1484–1488 CrossRef CAS.
- A. Seubert and G. Wünsch, Nebenreaktionen in Karl-Fischer-reagentien, Fresenius' Z. Anal. Chem., 1989, 334, 256–260 CrossRef CAS.
- E. R. Morris and J. C. J. Thynne, Reactions of radicals containing fluorine. Part 1. Hydrogen and deuterium atom abstraction from trideuteromethanol by trifluoromethyl radicals, Trans. Faraday Soc., 1968, 64, 414–421 RSC.
- J. Lillie, D. Behar, R. J. Sujdak and R. H. Schuler, Lifetime of trifluoromethyl radical in aqueous solution, J. Phys. Chem., 1972, 76, 2517–2520 CrossRef CAS.
- K. Seppelt, Trifluoromethanol, CF3OH, Angew. Chem., Int. Ed. Engl., 1977, 16, 322–323 CrossRef.
- M. Avataneo, U. De Patto, M. Galimberti and G. Marchionni, Synthesis of α,ω-dimethoxyfluoropolyethers: reaction mechanism and kinetics, J. Fluorine Chem., 2005, 126, 631–637 CrossRef.
- R. Flyunt, O. Makogon, M. N. Schuchmann, K.-D. Asmus and C. von Sonntag, OH-Radical-induced oxidation of methanesulfinic acid. The reactions of the methanesulfonyl radical in the absence and presence of dioxygen, J. Chem. Soc., Perkin Trans. 2, 2001, 787–792 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Potentiometric titration of 1b–1e irradiated solutions and 1H and 13C NMR spectra of compounds 1d–f. See DOI: 10.1039/c0pp00284d |
| ‡ Compound 2a, MS (m/z): 108 (M+, 100), 79 (15), 78 (60), 77 (20), 65 (35), 51 (10); 2a-d1, MS (m/z): 109 (M+), 80 (20), 79 (65), 78 (25), 66 (45), 52 (10); 4-d3, MS (m/z): 141 (M+), 126 (60), 123 (55), 98 (20), 95 (15). |
| This journal is © The Royal Society of Chemistry and Owner Societies 2011 |