The use of dendrimers as high-performance shells for round-trip energy transfer: efficient trans-cisphotoisomerization from an excited triplet state produced within a dendrimer shell
Received
7th September 2010
, Accepted 15th October 2010
First published on 9th November 2010
Abstract
A series of stilbene-cored poly(benzyl ether) dendrimers with benzophenone peripheries were synthesized and their photophysical and photochemical properties were studied. Fluorescence studies revealed that singlet–singlet energy transfer (SSET) from the stilbene core to the benzophenone units took place efficiently in dendrimers of all generations. Similarly, phosphorescence and time-resolved spectroscopic measurements indicated efficient triplet–triplet energy transfer (TTET) from the benzophenone periphery to the stilbene core. Upon excitation at 310 nm, the stilbene core isomerizes via an energy round trip within the dendrimer shell. The quantum yields for the energy round trip (ΦERT), defined as the product of the quantum yields of SSET, intersystem crossing, and TTET (ΦERT = ΦSSΦiscΦTT), were extremely high for all generations—99%, 95% and 94% for G1, G2, and G3, respectively—which means that the excitation energy of the dendrimer core was transferred to the dendrimer periphery and back to the core almost quantitatively. The quantum yield for photoisomerization of G1–G3via an energy round trip was higher than for other stilbene-cored dendrimers, which mainly isomerize from the excited singlet state. Photostability in the dendrimers was also demonstrated and discussed.
Introduction
Stilbene and its derivatives represent a large research area combining important branches of chemistry, biology and physics. In particular, their unique photochemical and photophysical features, including fluorescence and photoisomerization, have been the subject of extensive investigation for several decades.1–4
The photochemical and photophysical properties of stilbene can be controlled by the introduction of different aromatic ring or substituent groups which induce modification of the electronic environment. For example, the excited-state behaviors of ‘push-pull’ stilbenes5–11 indicate the important role of the intramolecular charge transfer or twisted intramolecular charge transfer state on deactivation. The photoreactivity of several monosubstituted hydroxystilbenes is highly dependent upon the position of the hydroxyl substituent.12,13 Replacing one of the phenyl rings with an anthracene ring causes a change in the potential energy surface, resulting in one-way cis-to-transphotoisomerization in both the excited singlet and triplet states.14,15
We also studied stilbenoid dendrimers, in which the stilbene acts as the core of the dendrimer, in order to investigate the effect of bulky dendron subunits on the excited-state properties.16,17 During the course of our dendrimer studies we found that a benzophenone-substituted stilbene (Fig. 1, G1) exhibited highly efficient intramolecular singlet–singlet energy transfer (SSET) from the excited stilbene to the benzophenone units and triplet–triplet energy transfer (TTET) from the excited triplet state of the benzophenone units to the stilbene. This was confirmed by quenching of fluorescence from the stilbene and phosphorescence from the benzophenone unit, respectively.18
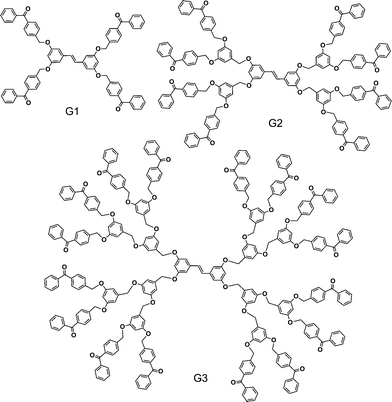 |
| | Fig. 1 Chemical structure of G1–G3dendrimers. | |
In this paper we report a unique intramolecular stilbene-benzophenone SSET–TTET system that was extended up to the G3 dendrimer (Fig. 1). Significantly, SSET took place more efficiently in G3 (98%) than in G2 (97%), and immediately after intersystem crossing in the benzophenone periphery, the triplet energy was flashed back to the stilbene core by TTET with greater than 96% efficiency in all generations. The stilbene core underwent photoisomerization from the triplet state in all generations. High photostability in the dendrimers was also demonstrated.
Experimental
Apparatus
Sample solutions were prepared in benzene and THF (Kanto Chemical) and deoxygenated by bubbling with highly purified argon (>99.999%) via a needle. Fluorescence and excitation spectra were measured on a Hitachi F-4500 fluorescence spectrophotometer with a 1 cm × 1 cm quartz cuvette. Absorption spectra were recorded using a Shimadzu UV-1600 spectrophotometer.
Fluorescence lifetimes were determined using a Horiba NAES-1100 time-resolved spectrofluorometer. Laser flash photolysis was performed using an excimer laser (Lambda Physik LPX-100, 308 nm, 20 ns fwhm) as an excitation light source and a pulsed xenon arc (Ushio UXL-159) as a monitoring light source. A photomultiplier (Hamamatsu R-928) and a storage oscilloscope (Iwatsu TS-123) were used for detection.
Materials
G1
The synthesis of G1 has been reported elsewhere.18
G2
G2-Br19 (0.37 g, 0.63 mmol), trans-3,3′,5,5′-tetrahydroxy-stilbene (30 mg, 0.12 mmol), 18-crown-6 ether (20 mg, 76 μmol) and K2CO3 (90 mg, 0.65 mmol) were reacted in THF (8 mL) and DMF (1 mL) under a nitrogen atmosphere, and the mixture was refluxed for 31 h. The reaction mixture was poured into water (100 mL) and extracted with dichloromethane (100 mL). The solvent was removed by evaporation under reduced pressure and the residue was purified by flash chromatography on silica gel using chloroform–hexane (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as an eluent. The resulting material was further purified by HPLC with a GPC column (TOSOH G2500HXL), using chloroform as an eluent, to give trans-G2 as a pale yellow amorphous powder (50 mg, 22 μmol, 18%). δH(400 MHz; CDCl3, Me4Si) 5.01 (8H, s, stil-O-CH2), 5.13 (16H, s, BP-CH2-), 6.52 (2H, t, J = 2.1 Hz, stil-Ph-4.4′), 6.59 (4H, t, Ph-4), 6.71 (8H, d, Ph-2,6), 6.74 (4H, d, J = 2.4 Hz, stil-Ph-2,6,2′,6′), 6.98 (2H, s, –CH
:1) as an eluent. The resulting material was further purified by HPLC with a GPC column (TOSOH G2500HXL), using chloroform as an eluent, to give trans-G2 as a pale yellow amorphous powder (50 mg, 22 μmol, 18%). δH(400 MHz; CDCl3, Me4Si) 5.01 (8H, s, stil-O-CH2), 5.13 (16H, s, BP-CH2-), 6.52 (2H, t, J = 2.1 Hz, stil-Ph-4.4′), 6.59 (4H, t, Ph-4), 6.71 (8H, d, Ph-2,6), 6.74 (4H, d, J = 2.4 Hz, stil-Ph-2,6,2′,6′), 6.98 (2H, s, –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–), 7.58–7.43 (40H, m, BP-3′,5′,3,4,5), 7.81–7.76 (32H, m, BP-2,6,2′,6′). δC (100 MHz, CDCl3, MeSi4) 69.5, 69.9, 101.7, 105.9, 106.6, 127.0, 128.3, 129.3, 130.0, 130.4, 132.5, 137.2, 137.5, 139.2, 139.5, 141.4, 159.9, 160.0, 196.2. MALDI-TOF-MSm/z: [M + Na]+ calcd for C154H116O20Na, 2307.80; found 2309.10.
CH–), 7.58–7.43 (40H, m, BP-3′,5′,3,4,5), 7.81–7.76 (32H, m, BP-2,6,2′,6′). δC (100 MHz, CDCl3, MeSi4) 69.5, 69.9, 101.7, 105.9, 106.6, 127.0, 128.3, 129.3, 130.0, 130.4, 132.5, 137.2, 137.5, 139.2, 139.5, 141.4, 159.9, 160.0, 196.2. MALDI-TOF-MSm/z: [M + Na]+ calcd for C154H116O20Na, 2307.80; found 2309.10.
G3
.
G3-Br8 (0.68 g, 0.55 mmol), trans-3,3′,5,5′-tetrahydroxy-stilbene (24 mg, 0.098 mmol), 18-crown-6 ether (20 mg, 76 μmol) and K2CO3 (100 mg, 0.7 mmol) were reacted in THF (6 mL) and DMF (3 mL) under a nitrogen atmosphere, and the mixture was refluxed for 20 h. The reaction mixture was poured into water (100 mL) and extracted with chloroform (100 mL × 4). The solvent was removed by evaporation under reduced pressure, and the residue was purified by flash chromatography on silica gel using chloroform/ether (30:1) as an eluent. The resulting material was further purified by HPLC with a GPC column (TOSOH G2500HXL), using chloroform as an eluent, to give trans-G3 as a pale yellow amorphous powder (200 mg, 42 μmol, 43%). δH(400 MHz; CDCl3; Me4Si) 4.94 (24H, s, –O–CH2-Ph & Stil-O-CH2), 5.06 (32H, s, –O–CH2-BP), 6.49 (2H, t, J = 2.1 Hz, stil-Ph-4.4′), 6.52 (4H, t, J = 2.1 Hz, Ph), 6.52 (8H, t, J = 2.1 Hz, Ph), 6.65–6.67 (24H, m, Ph), 6.78 (4H, d, J = 2.0 Hz, stil-Ph-2,6,2′,6′), 6.94 (2H, s, –CHCH–), 7.55–7.40 (80H, m, BP-3,4,5,3′,5′), 7.77–7.73 (64H, m, BP-2,6,2′,6′). δC (100 MHz; CDCl3; MeSi4) 69.4, 69.9, 69.8, 101.6, 101.7, 106.0, 106.4, 106.5, 127.0, 128.3, 129.2, 130.0, 130.4, 132.5, 137.1, 137.4, 139.2, 139.3, 139.4, 141.3, 159.9, 160.0, 196.2. MALDI-TOF-MSm/z: [M + Na]+ calcd for C322H244O44Na, 4836.68; found, 4836.82.
Results and discussion
1. Singlet-singlet energy transfer from core to periphery
1.1.
Absorption spectra
.
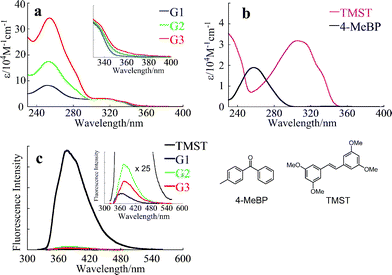
The dendrimers consisted of a stilbene core, benzyl ether branching units, and peripheral benzophenones, and their absorption spectra were a superposition of the band for each unit. Fig. 2 shows the absorption spectra of the G1–G3dendrimers (Fig. 2a), and their model compounds, trans-tetramethoxystilbene (TMST), and 4-methylbenzophenone (4-MeBP) (Fig. 2b) measured in THF. The bands around 300–350 nm in Fig. 2a were mainly due to the π–π* transition of the stilbene core, with almost the same the band shapes and intensities among all dendrimers, while the band at 250–290 nm increased in intensity in the higher generations because it was due to a combination of branching units and peripheral benzophenones. The ε values of the dendrimers at 252 nm were as large as 174000 M−1 cm−1 for G2 and 345000 M−1 cm−1 for G3, respectively, showing a significant antenna effect in the higher generations (Table 1). The small shoulder around 360–390 nm (Fig. 2a, inset), increasing in the higher generations, was assigned to the n–π* transition of the benzophenones. The band shapes and the location of each dendrimer unit were similar to those of the reference compounds TMST and 4-MeBP, which indicated that there was almost no electronic interaction between the stilbene and the benzophenone units in the ground state. The stilbene π–π* band appeared at a shorter wavelength than the benzophenone n–π* absorption band, suggesting that singlet–singlet energy transfer (SSET) from the excited stilbene (83 kcal mol−1)20 to the benzophenone units (72 kcal mol−1)21 is exothermic.
Table 1 Absorption maxima, molar extinction coefficients, fluorescence quantum yields, fluorescence lifetimes, and singlet–singlet energy transfer quantum yields and rate constants for TMST and G1–G3dendrimers
| |
λabs/nm (ε/M−1 cm−1)a |
Φfb |
τs/nsb,c |
ΦSS |
k
SS/1010s−1 |
|
Measured in THF.
Measured in benzene.
Estimated from eqn (1).
Ref. 5.
Ref. 18.
|
|
TMST
d |
309 (31900) |
0.24 |
4.1 |
— |
— |
| G1e |
251 (83000) |
0.002 |
0.034 |
0.99 |
2.92 |
| |
310 (31000) |
|
|
|
|
| G2 |
252 (174000) |
0.007 |
0.131 |
0.97 |
0.74 |
| |
310 (29500) |
|
|
|
|
| G3 |
253 (345000) |
0.004 |
0.076 |
0.98 |
1.30 |
| |
310 (30200) |
|
|
|
|
1.2.
Fluorescence spectra
.
The fluorescence spectra of the dendrimers and TMST were measured in benzene at room temperature (Fig. 2c). Strong fluorescence (λmax = 373 nm, Φf = 0.24 in benzene)5 was observed for TMST on excitation near the maximal absorption (310 nm). Upon excitation at the same wavelength, all of the dendrimers exhibited fluorescence spectra similar to that of TMST, but with very weak intensities (Fig. 2c, inset), probably due to intramolecular S–S energy transfer from the excited stilbene to the surrounding benzophenone units, leading to quenching of fluorescence emissions. The fluorescence quantum yields (Φf) of the dendrimers were determined to be 0.002, 0.007 and 0.004 for G1, G2 and G3, respectively, almost two orders of magnitude less than TMST. In particular, the Φf of G3 was less than that of G2, suggesting that energy transfer takes place more efficiently in G3 than in G2.
1.3. Fluorescence lifetimes.
To estimate the efficiency of SSET (ΦSS) from the core stilbene to the peripheral benzophenones, the fluorescence lifetimes (τs) were measured by the single photon counting method in benzene under argon. However, the τs values of all dendrimers were found to be less than 0.5 ns, which was the equipment limit. Therefore, more precise τs values were estimated based on eqn (1), in which the value of the fluorescence emission rate constant (kf) of the model compound TMST was used to obtain a best estimate:Furthermore, it was possible to estimate the rate constant of SSET (kSS) and the efficiency (ΦSS) based on eqn (2) and (3), respectively:| | | kSS = 1/τs − (kf + kiso + knr) | (2) |
where the rate constants of photoisomerization (kiso) and non-radiative decay (knr) in eqn (2) are the values for TMST. The estimated kSS values for the dendrimers were very large (G1: kSS = 2.9 × 1010 s−1; G2: kSS = 7.4 × 109 s−1; G3: kSS = 1.3 × 1010 s−1); consequently, singlet–singlet energy transfer was expected to take place with high quantum yield (ΦSS = 0.99, 0.97 and 0.98 for G1, G2 and G3, respectively). Significantly, the energy transfer efficiency in G3 seemed to be higher than in G2. Because of the flexibility of the benzyl ether dendron, the distance between the stilbene and the closest benzophenone units in G3 was expected to be smaller than in G2. We calculated the lowest energy conformation for each generation using the Spartan 04 program. In this case the solvent effect was not considered. The distances between the carbonyl C of the benzophenone unit and the closest C in the CC double bond of stilbene are shown in Table 2; these were in agreement with data obtained by others.19,22 The average donor–acceptor distances were longer in higher generations (G3: 17.3 Å, Table 2) but the shortest donor–acceptor distances were also observed in G3. SSET took place with extremely high quantum yield in all generations because it proceeds by the Förster (through-space) mechanism, for which the effective radius is about 50 Å.23 In view of the fact that the ΦSS value of G3 was higher than that of G2, SSET may be partly associated with the number of energy acceptor benzophenones.
Table 2 Donor–acceptor distances for G1–G3dendrimers
| Donor–acceptor distance/Å |
G1 |
G2 |
G3 |
| Shortest |
7.45 |
6.54 |
6.34 |
| Average |
9.82 |
13.6 |
17.3 |
2. Triplet–triplet energy transfer from periphery to core
2.1.
Phosphorescence spectra
.
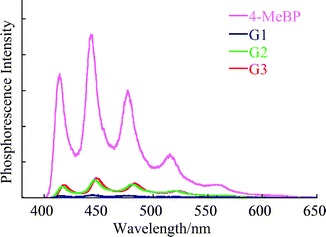
When the excitation energy is transferred to the benzophenone unit, it immediately undergoes intersystem crossing to the excited triplet state.24 To examine this, the phosphorescence spectra of the dendrimers and the reference compound 4-MeBP were studied in a 2-methyltetrahydrofuran (M-THF) rigid matrix at 77 K under argon. The phosphorescence spectrum of 4-MeBP under light irradiation at 370 nm showed strong vibrational structures at 414, 443, and 476 nm. Similar structures were detected in the phosphorescence spectra from all dendrimers (Fig. 3), but their intensities were much weaker than those of 4-MeBP. These results indicate that the excited triplet benzophenone units in the dendrimers were efficiently quenched by the core stilbene even in higher generations. In other words, there was efficient intramolecular triplet–triplet energy transfer (TTET) from the benzophenone units to the stilbene core. In fact, based on the triplet-state energies of the benzophenone units in the dendrimers (69.2, 68.7, and 68.4 kcal mol−1 for G1, G2, and G3, respectively), the TTET was exothermic. The triplet energy of TMST was 49 kcal mol−1.25 The quantum yield for intramolecular TTET (ΦTT) was estimated by comparing the phosphorescence intensity of the dendrimers with that of 4-MeBP, giving 0.99, 0.88, and 0.86 for G1, G2, and G3, respectively; this shows a dependence on the dendrimer generation. In general, an electron exchange (Dexter) mechanism is accepted for TTET:26| | | n(l) = (1/ô0)exp[2(R0 − l)/L] | (4) |
where l is the interchromophore distance, ô0 is the lifetime of the triplet excited state, R0 is an interchromophore distance such that the probability of relaxation to the ground state is the same as that of energy transfer, and L is a constant called the effective average Bohr radius.27 The effective distance for the Dexter mechanism is less than 10 Å.26 According to eqn (4) for the Dexter mechanism, triplet–triplet energy transfer depends critically on the distance between the donor and the acceptor. The ΦTT values, therefore, indicate that the average distance between the stilbene and benzophenone units is greater in higher generations of the dendrimer, at least in the rigid matrix at 77 K.
2.2. Transient absorption spectra.
To gain more insight into intramolecular TTET in dendrimers, flash photolysis was performed using a 308 nm excimer laser in benzene under argon at 298 K. Typical transient absorption spectra for the model compound 4-MeBP and the G3dendrimer are shown in Fig. 4. The absorption around 530 nm in Fig. 4a was attributed to the T-T absorption band for the lowest triplet state of the benzophenone, which was rapidly quenched by oxygen. A transient absorption was observed in the G3dendrimer (Fig. 4b) in the same wavelength region, but the intensity was significantly weaker than the equivalent absorption in 4-MeBP. The time profile of the transient absorption at 530 nm is shown in Fig. 4c and the set of acquired data is summarized in Table 3. The triplet lifetimes were determined to be 2.5 μs for 4-MeBP, < 20 ns (less than the equipment limit) for G1, and 60 and 90 ns for G2 and G3, respectively.
Table 3 Triplet lifetimes and TTET quantum yields and rate constants for 4-MeBP and G1–G3dendrimers
| |
τT/μsa |
ΦTT |
k
TT
/107s−1 |
| 77 Kb |
298 Ka |
|
Measured in benzene at 298 K.
Measured in 2-MTHF.
|
|
4-MeBP
|
2.5 |
— |
— |
— |
| G1 |
<0.02 |
0.99 |
0.99 |
4.96 |
| G2 |
0.06 |
0.88 |
0.98 |
1.63 |
| G3 |
0.09 |
0.86 |
0.96 |
1.07 |
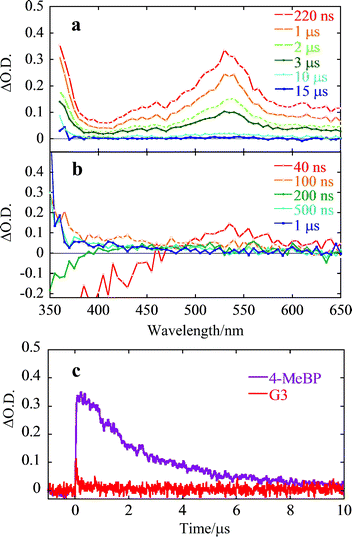 |
| | Fig. 4 Transient absorption spectra of (a) 4-MeBP and (b) G3dendrimer, and (c) decay curves for transient species at 530 nm for 4-MeBP and G3. | |
The triplet lifetime increased as the dendrimer generation increased. The shorter triplet-state lifetimes of the dendrimers and the generation-dependent trend are consistent with the results of the phosphorescence experiments and strongly indicate the occurrence of intramolecular TTET from the triplet benzophenone units to the stilbene core. The rate constant for TTET (kTT) and its quantum yield (ΦTT) at 298 K can be calculated from the triplet lifetime of Gn and 4-MeBP according to eqn (5), and (6) respectively. The results are summarized in Table 3.
The kTT values can be expected to follow the order of kSS, because kSS will lead the preferential population of the triplet at the singlet-energy-accepted benzophenone due to very fast intersystem crossing. However, the actual kTT values follow the average donor–acceptor distances (Table 2). This results indicate a competition between kTT and other process like triplet energy hopping that can scatter the triplet energy to the peripheral benzophenones.
The decrease in the ΦTT value with increasing generations was consistent with the tendency demonstrated by phosphorescence measurement at 77 K, but the efficiencies at 298 K were higher than those measured at 77 K for the same generation. More importantly, the ΦTT values were quite high even in the G3dendrimer (ΦTT = 0.96). Based on a TTET system using the same benzophenone dendrons as in the current work, Liet al. reported ΦTT values of 0.98 for G1 but only 0.56 for G2; it was concluded that the reason for the low ΦTT values was not only the longer distance between the donor and the acceptor but also triplet–triplet annihilation in higher generations.19 The difference between Liet al.'s dendrimers and ours may be morphological. In our system, the core stilbene is spherically covered by four dendrons. It was reported previously that a tetra-substituted porphyrin dendrimer with a spherical morphology acted as an efficient light-harvesting antenna, with the excitation energy becoming delocalized and migrating over the dendrimeric array surrounding the singlet energy acceptor, resulting in enhanced energy transfer efficiency.28 Although there are no reports dealing with the morphology of triplet state dendrimers, the aforementioned findings are a clear example of the effect of dendrimer morphology on the efficiency of triplet–triplet energy transfer.
In addition, considering the duration of the laser pulse and fast SSET and intersystem crossing in benzophenone, it can be possible to form several triplet benzophenone in a single dendrimer that may lead to T-T annihilation24via triplet energy hopping between different benzophenones.
3.
Photoisomerization by round-trip excitation energy
The photosensitized trans-cisisomerization of stilbene (intermolecular or intramolecular) has been the subject of many studies over the last 40 years.29,30 In most cases, benzophenone is selectively excited to produce excited triplet stilbene;25 however, in our system, stilbene can be directly excited to produce triplet stilbene. After excitation, singlet energy transfer to the peripheral benzophenone units occurs with extremely high quantum yield. The benzophenone units then undergo intersystem crossing, and the triplet energy comes back to the stilbene core with a quantum yield of 0.96–0.99 at 298 K. Thus, the quantum yield of this round-trip energy transfer (ΦERT) was estimated based on eqn (7).where Φisc represents the quantum yield of intersystem crossing in the benzophenone units and can be assumed to be quantitative (Φisc = 1).24 The ΦERT values for all dendrimers were calculated to be as high as 0.94–0.99 (Table 4). In this case, the production of triplet stilbene by direct intersystem crossing in stilbene itself is thought to be negligible due to the fact that the rate constant for SSET (kSS = 1010s−1) is probably much greater than those of other photochemical processes.5 The round-trip energy transfer, depicted in Fig. 5, ends with the photochemical trans-cisisomerization of triplet stilbene.
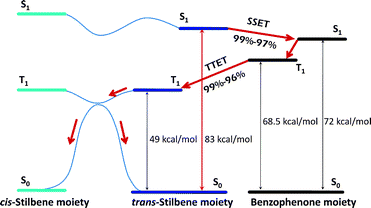 |
| | Fig. 5 Energy diagram for photoreaction of G1–G3dendrimers. | |
Table 4 Quantum yield for energy round trip, quantum yield for trans-cisphotoisomerization, populations of trans and cis-isomers in the photo-stationary state, and photosensitivity at 252 nm for G1–G3dendrimers
| |
ΦERT |
Φiso |
trans/cis (pss) |
| G1 |
0.99 |
0.47 |
45/55 |
| G2 |
0.95 |
0.38 |
55/45 |
| G3 |
0.94 |
0.41 |
45/55 |
Based on the mechanism described above, light excitation of the dendrimers at 310 nm resulted in trans-cisphotoisomerization, as revealed by the change in the absorption spectrum (Fig. 6). Under these conditions, only the stilbene core absorbs light (Fig. 2b). The Φt→c values were determined as 0.47, 0.38 and 0.41 for G1, G2 and G3, respectively. The apparent Φt→c values can be considered to be one half of that of CC bond rotation in the excited state (Fig. 5), and Φt→c values for stilbene are usually a maximum of 0.5.15
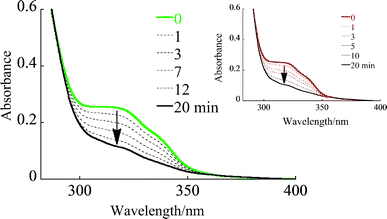 |
| | Fig. 6 Change in the absorption spectra of G3 and (inset) G2dendrimers upon light irradiation at 310 nm. | |
Although the isomerization was accomplished via three processes, including two energy transfers and intersystem crossing, the quantum yields for the dendrimers were still quite high. In fact, the Φt→c values for G1–G3 were higher than for other stilbene-cored dendrimers,16 which undergo isomerization from the excited singlet state. This is probably because photoisomerization competes with other deactivation processes in the excited singlet state of SDs, while in the triplet state, stilbene is mainly deactivated through CC bond rotation.31 The molar ratio of trans and cis isomers at the photostationary state ([t]/[c]pss) was 50:50, which was different from the value obtained for isomerization from the excited singlet state.
Finally, we demonstrated “typical” photosensitized isomerization. Upon irradiation with light at 370 nm, G1–G3 showed changes in their absorption spectra due to trans-cisisomerization. Under these conditions, only the benzophenone units absorb light; thus, isomerization must be caused by T-T energy transfer. An isosbestic point was observed during isomerization (data not shown), indicating that the photosensitized reaction is a remarkably clean process. In other words, rapid T-T energy transfer in the excited benzophenones is prioritized over hydrogen abstraction or T-T annihilation, which are usually observed in other energy transfer systems.19,24 Moreover, photocyclization of cis-stilbene to give dihydrophenanthrene was not observed, because cyclization proceeds via the excited singlet state.20
4. Photostability
As mentioned above, benzophenone is known to undergo efficient hydrogen abstraction in the excited triplet state. The details of the H abstraction reaction for triplet benzophenone have been studied extensively.24 In the G2 and G3dendrimers, benzyl groups on the branching units might be expected to act as H donors; therefore, there is a possibility that intramolecular H abstraction could occur in excited triplet benzophenones, leading to decomposition of the dendrimer. However, irradiation of G2 and G3 with light at 370 nm in deaerated benzene solution, with the irradiation time extended to more than 180 min, led only to a 1:1 mixture of trans- and cis-isomers, with no detectable change in the absorption spectra. This result indicates that T-T energy transfer from the benzophenone units to the stilbene core is much faster than other photochemical reactions, including intramolecular H abstraction. The photostability of G1 has been reported elsewhere.18
5. Efficient Dexter-type (spin exchange) triplet energy transfer at long donor–acceptor distances
This report demonstrates clearly that intramolecular energy transfer processes took place very efficiently in dendritic molecules, even in the case of triplet energy transfer with an average donor–acceptor distance of as much as 1.5 nm. The energy transfer rate constant may be estimated to be as high as ca. 107 mol−1 dm3s−1, based on the quenching of the benzophenone triplet state by the stilbene core, which results in trans-cisisomerization in the triplet state.
Usually, triplet energy transfer with an electron exchange process requires a collision between the donor molecule and the acceptor molecule, and the efficiency decreases dramatically with increasing distance. However, in the dendritic molecules used in this study, the energy transfer rate constant remained quite high. To the best of our knowledge, this is the first clear evidence of the occurrence of triplet energy transfer between donor and acceptor at considerable distance, inducing highly efficient trans-cisisomerization in the excited triplet state. As the stilbene chromophore absorbs more intensely than benzophenone at 300–350 nm, the round-trip energy transfer process strongly enhances the light absorption efficiency of the dendrimer using the light for excitation of the stilbene chromophore. In other words, self-sensitization—involving light absorption by a reactant with a high extinction coefficient, followed by S–S energy transfer, intersystem crossing, and T–T energy transfer—results in efficient production of the reactive triplet state. This process could be used as a highly efficient light-harvesting system for triplet-sensitized reactions which cannot usually be achieved by direct excitation of the reacting chromophore.
Conclusions
Efficient and clean photochemical trans-cisisomerization by round-trip energy transfer was demonstrated in stilbene-cored dendrimers with a benzophenone periphery. The energy round trip was initiated by singlet–singlet energy transfer from the stilbene core to the benzophenone periphery, followed by intersystem crossing in the benzophenone, and then triplet–triplet energy transfer from the benzophenone units back to the stilbene core. The quantum yields for the round-trip energy transfer were extremely high, even in the G3dendrimer (ΦERT = 0.94–0.99). In addition, although several processes took place, the quantum yields for photoisomerization were still high (Φt→c = 0.38–0.47). These findings reveal that an efficient energy transfer system involving an antenna chromophore may be used to harvest photon energy in order to initiate chemical reactions that proceed in the excited triplet state.
Acknowledgements
This work was supported by Grants-in-Aid for Scientific Research in a Priority Area, “New Frontiers in Photochromism” (Nos. 471 and 19550176), from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT), Japan.
Notes and references
-
J. Saltiel, J. D'Agostino, E. D. Megarity, L. Metts, K. R. Neuberger, M. Wrighton and O. C. Safiriou, in Organic Photochemistry, ed. O. L. Chapman, Marcel Dekker, New York, 1973, vol. 3, p. 1 Search PubMed.
-
J. Saltiel and J. L. Charlton, in Rearrangements in Ground and Excited States, ed. P. Mayo, Academic Press, New York, 1980, vol. 3, p. 25 Search PubMed.
- D. H. Waldeck, Chem. Rev., 1991, 91, 415 CrossRef CAS.
-
G. Likhtenshtein, in Stilbenes: Applications in Chemistry, Life Sciences and Materials Science, Wiley-VCH, Weinheim, Germany, 2010 Search PubMed.
- J. Hayakawa, M. Ikegami, T. Mizutani, M. Wahadoszamen, A. Momotake, Y. Nishimura and T. Arai, J. Phys. Chem. A, 2006, 110, 12566 CrossRef CAS.
- H. Gruen and H. Görner, J. Phys. Chem., 1989, 93, 7144 CrossRef CAS.
- R. Lapouyade, A. Kuhn, J. F. Letard and W. Rettig, Chem. Phys. Lett., 1993, 208, 48 CrossRef CAS.
- N. Eilers-König, T. Kühne, D. Schwarzer, P. Vöhringer and J. Schoroeder, Chem. Phys. Lett., 1996, 253, 69 CrossRef.
- V. Papper, D. Pines, G. Likhtenshtein and E. Pines, J. Photochem. Photobiol., A, 1997, 111, 87 CrossRef CAS.
- F. D. Lewis and W. Weigel, J. Phys. Chem. A, 2000, 104, 8146 CrossRef CAS.
- J.-S. Yang, K.-L. Liau, C.-Y. Hwang and C.-M. Wang, J. Phys. Chem. A, 2006, 110, 8003 CrossRef CAS.
- T. Murohoshi, K. Kaneda, M. Ikegami and T. Arai, Photochem. Photobiol. Sci., 2003, 2, 1247 RSC.
-
(a) F. D. Lewis and E. M. Crompton, J. Am. Chem. Soc., 2003, 125, 4044 CrossRef CAS;
(b) E. M. Crompton and F. D. Lewis, Photochem. Photobiol. Sci., 2004, 3, 660 RSC.
- T. Arai, H. Karatsu, H. Sakuragi and K. Tokumaru, Tetrahedron Lett., 1983, 24, 2873 CrossRef CAS.
- T. Arai and K. Tokumaru, Chem. Rev., 1993, 93, 23 CrossRef CAS.
-
(a) M. Uda, T. Mizutani, J. Hayakawa, A. Momotake, M. Ikegami, R. Nagahata and T. Arai, Photochem. Photobiol., 2002, 76, 596 CAS;
(b) S. Watanabe, M. Ikegami, R. Nagahata and T. Arai, Bull. Chem. Soc. Jpn., 2007, 80, 586 CrossRef CAS;
(c) J. Hayakawa, A. Momotake and T. Arai, Chem. Commun., 2003, 94 RSC.
-
(a) T. Okamoto, A. Momotake, Y. Shinohara, R. Nagahata and T. Arai, Bull. Chem. Soc. Jpn., 2007, 80, 2226 CrossRef CAS;
(b) C. Mitsuno, A. Momotake, Y. Shinohara, K. Takahashi, R. Nagahata, Y. Nishimura and T. Arai, Tetrahedron Lett., 2009, 50, 7074 CrossRef CAS.
- Y. Miura, A. Momotake, Y. Shinohara, M. Wahadoszamen, Y. Nishimura and T. Arai, Tetrahedron Lett., 2007, 48, 639 CrossRef CAS.
- J. Chen, S. Li, L. Zhang, B. Liu, Y. Han, G. Yang and Y. Li, J. Am. Chem. Soc., 2005, 127, 2165 CrossRef CAS.
- A. Momotake, M. Uda and T. Arai, J. Photochem. Photobiol., A, 2003, 158, 7 CrossRef CAS.
- J. Saltiel, H. C. Curtis, L. Metts, J. W. Miley, J. Winterle and M. Wrighton, J. Am. Chem. Soc., 1970, 92, 410 CrossRef CAS.
- A. Adronov, S. L. Gilat, J. M. J. Fréchet, K. Ohta, F. V. R. Neuwahl and G. R. Fleming, J. Am. Chem. Soc., 2000, 122, 1175 CrossRef CAS.
- T. Förster, Ann. Phys., 1948, 437, 55 CrossRef.
-
N. J. Turro, Modern Molecular Photochemistry, Benjamin/Cummings, Menlo Park, CA, 1978, chapter 9 Search PubMed.
- H. Görner and D. Schulte-Frohlinde, J. Phys. Chem., 1981, 85, 1835 CrossRef.
- D. L. Dexter, J. Chem. Phys., 1953, 21, 836 CrossRef CAS.
- M. Inokuti and F. Hirayama, J. Chem. Phys., 1965, 43, 1978 CrossRef CAS.
- D.-L. Jiang and T. Aida, J. Am. Chem. Soc., 1998, 120, 10895 CrossRef.
-
(a) G. S. Hammond, J. Saltiel, A. A. Lamola, N. J. Turro, J. S. Bradshaw, D. D. Cowan, R. C. Counsell, V. Vogt and C. J. Dalton, J. Am. Chem. Soc., 1964, 86, 3197 CrossRef CAS;
(b) H. Görner and D. Schulte-Frohlinde, J. Phys. Chem., 1979, 83, 3107 CrossRef;
(c) J. Saltiel, S. Ganapathy and C. Werking, J. Phys. Chem., 1987, 91, 2755 CrossRef CAS.
- M. R. Ams and C. S. Wilcox, J. Am. Chem. Soc., 2006, 128, 250 CrossRef CAS; M. R. Ams and C. S. Wilcox, J. Am. Chem. Soc., 2007, 129, 3966 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry and Owner Societies 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.