Luminescence of meso-pyrimidinylcorroles: relationship with substitution pattern and heavy atom effects
Francesco
Nastasi
a,
Sebastiano
Campagna
*a,
Thien H.
Ngo
b,
Wim
Dehaen
*b,
Wouter
Maes
bc and
Mikalai
Kruk
*d
aDipartimento di Chimica Inorganica, Chimica Analitica e Chimica Fisica, Università di Messina, and Centro Interuniversitario per la Conversione Chimica dell'Energia Solare, via Sperone 31, 98166, Messina, Italy
bMolecular Design and Synthesis, Department of Chemistry, Katholieke Universiteit Leuven, Celestijnenlaan 200F, B-3001, Leuven, Belgium
cInstitute for Materials Research (IMO), Research Group Organic and (Bio)Polymer Chemistry, Hasselt University, Universitaire Campus, Agoralaan Building D, B-3590, Diepenbeek, Belgium
dB.I. Stepanov Institute of Physics of National Academy of Sciences, Pr. Nezavisimosti, 68, 220072, Minsk, Belarus
First published on 19th November 2010
Abstract
The luminescence properties of a series of corroles containing multiple meso-pyrimidinyl groups have been studied. In particular, nine corroles containing two pyrimidinyl moieties, four corroles containing three pyrimidinyl groups and one corrole carrying a single pyrimidinyl substituent have been investigated, and their properties have been compared with some model species. The results indicate that the energy of the emissive π–π* corrole-core-based state is not significantly perturbed by the various substituents, whereas the emission lifetimes and quantum yields depend on the number of appended meso-dichloropyrimidinyl substituents. In particular, both emission lifetime and quantum yield decrease with increasing the number of meso-dichloropyrimidinyl substituents, whereas pyrimidinyl substituents which do not carry further electron withdrawing groups such as the chlorine atoms do not affect the corrole emission properties. Two hypotheses are taken into consideration to rationalize the results: (i) the presence of meso-dichloropyrimidinyl substituents could introduce low-lying CT states which mix with the corrole π–π* emissive level, so reducing emission efficiency; (ii) the ortho,ortho′-chlorine groups of the meso-pyrimidinyl substituents, which lie in the proximity of the corrole macrocycle, can increase the intersystem crossing rate constant of the corrole-based fluorescent state via the heavy-atom effect. A comparison of the results of the studied corrole compounds with formerly investigated species and the linear dependence of the radiationless decay rate constants and emission quantum yields on the squared spin–orbit coupling constants, calculated considering the number of chlorine atoms in ortho-position of the corrolemeso-substituents, suggest that hypothesis (ii) is most likely the valid one. These results are of particular interest for the design of (a) corrole compounds featuring highly efficient triplet formation and (b) multicomponent systems containing photo-active corrole subunits and pyrimidinyl spacers.
Introduction
Corroles are extensively studied since their ground and excited state properties make them quite interesting for several theoretical and applied reasons, such as catalysis, sensor design, molecular electronics, solar energy conversion, medical imaging and therapy.1,2 Quite recently, corroles have also been included in more elaborate macromolecular systems, taking advantage of synthetic progress,3 in particular as far as suitable functionalization of corrole structures is concerned, which opened the way for the connection of corroles with other photo- and/or redox-active systems.4 Among the different approaches to corrole functionalization towards corrole-based building blocks for multicomponent systems, the preparation of meso-pyrimidinyl-substituted corroles has been successfully explored by our team.5,6We have recently investigated AB2-corroles containing one pyrimidinyl substituent bearing different attached groups,6 and have demonstrated that the photophysical properties of the corrole core are significantly affected by the presence of chlorine atoms as ortho,ortho′-substituents on the pyrimidinyl group (hereafter, dichloropyrimidinyl substituent). Here we report on the luminescence properties of a large series of A2B- and A3-pyrimidinylcorroles containing either two or three meso-pyrimidinyl groups bearing different attached groups, to evaluate the effect of multiple (dichloro)pyrimidinyl moieties on the photophysical properties of the corrole macrocycle. In particular, the luminescence properties of nine corroles containing two pyrimidinyl units (compounds 1–7, 11, and 13) and four corroles containing three pyrimidinyl units (compounds 9, 10, 12, and 14) are reported. The properties of a species having a single pyrimidinyl group (compound 8) are also investigated. Fig. 1 shows the structural formulae of all the corroles studied here, together with those of a couple of recently investigated compounds (A and B), used as reference samples.
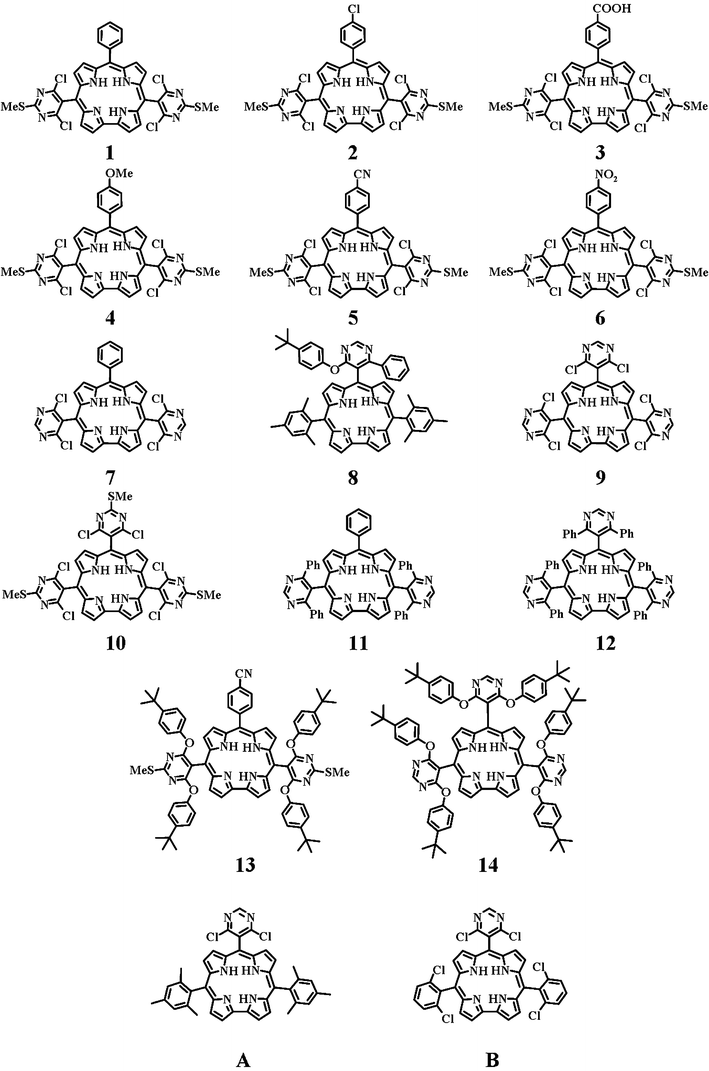 | ||
| Fig. 1 Structural formulae of the studied compounds. | ||
Results and discussion
Absorption spectra
The absorption spectra of all the corroles studied are dominated by bands which are attributed to spin-allowed π–π* transitions of the conjugated corrole macrocycle analogous to those of porphyrins, with intense Soret band(s) around 420 nm and moderately intense Q bands in the range 500–700 nm.1e,2,4Fig. 2 shows the absorption spectra of representative compounds and Table 1 collects the relevant spectral features of all the species.| Corrole | Absorption | Luminescence, 298 K | ||||
|---|---|---|---|---|---|---|
| λmax/nm (ε/M−1 cm−1) | λmax/nm | τ/ns | Φ | k r/107s−1 | k nr/108s−1 | |
| 1 | 416 (135000); 575 (22000); 618 (13500); 645 (8000) | 655 | 1.25 | 0.040 | 3.20 | 7.68 |
| 2 | 418 (145000); 575 (25000); 612 (15000); 640 (6500) | 655 | 1.42 | 0.036 | 2.54 | 6.79 |
| 3 | 416 (120000); 570 (23500); 618 (15000); 645 (10000); | 655 | 1.35 | 0.020 | 1.48 | 7.26 |
| 4 | 417 (127000); 570 (20000); 617 (13500); 645 (9000) | 665 | 1.23 | 0.038 | 3.09 | 7.82 |
| 5 | 416 (146500); 570 (25500); 612 (15000); 640 (6500) | 655 | 1.41 | 0.030 | 2.13 | 6.88 |
| 6 | 420 (115500); 570 (24000); 612 (13000); 640 (5300) | 660 | 0.42 | 0.011 | 2.62 | 23.55 |
| 7 | 413 (115000); 565 (16500); 615 (11500); 640 (7000) | 660 | 1.32 | 0.029 | 2.20 | 7.36 |
| 8 | 412 (113000); 428 (96000); 570 (18500); 603(11000); 635(4500) | 650 | 4.81 | 0.132 | 2.74 | 1.80 |
| 9 | 415 (123000); 575 (20000); 614 (9500); 640(2200) | 650 | 0.77 | 0.013 | 1.69 | 12.82 |
| 10 | 420 (132000); 575 (20000); 612 (10500); 640 (2500) | 650 | 0.75 | 0.016 | 2.13 | 13.12 |
| 11 | 425 (120000); 588 (20000); 625 (13500); 655 (7500) | 675 | 4.11 | 0.06 | 1.46 | 2.29 |
| 12 | 432 (114000); 595 (17500); 630 (11500); 660 (6200) | 670 | 4.32 | 0.10 | 2.31 | 2.08 |
| 13 | 420 (135000); 580 (25000); 615 (16500); 645 (8500) | 665 | 4.3 | 0.11 | 2.56 | 2.07 |
| 14 | 417 (132000); 575 (24000); 614 (14000); 640 (6400) | 660 | 4.42 | 0.13 | 2.94 | 1.97 |
| A | 412 (107300); 428 (89000); 570 (2090); 600 (11 200); 635 (3370) | 647 | 2.43 | 0.066 | 2.72 | 3.84 |
| B | 415 (126800); 570 (25000); 608 (13100) | 650 | 0.8 | 0.020 | 2.50 | 12.25 |
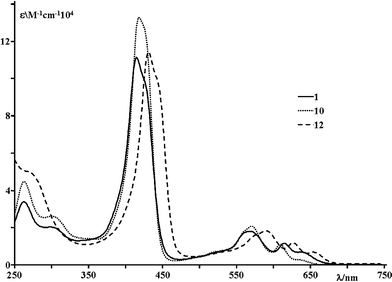 | ||
| Fig. 2 Absorption spectra of 1, 10 and 12 in dichloromethane solution. | ||
The noticeable features of the absorption are the splitting of the Soret band, analogous to the findings reported for other investigated corroles with sterically demanding meso-substituents,7 and the slight red shift (apparent in both Soret and Q bands) exhibited by compounds 11 and 12 (see Fig. 2). This red shift is probably due to slight perturbations of the corrole core orbitals as a consequence of sterical effects involving the phenyl groups directly linked as ortho-groups to the pyrimidinyl substituents and therefore spatially close to the corrole core orbitals.
Luminescence properties
All the compounds examined exhibit a relatively intense, structured emission, with lifetimes decaying within the nanosecond timescale. Fig. 3 shows representative luminescence spectra and relevant photophysical data are gathered in Table 1. The luminescence properties are independent of excitation wavelength, both exciting in the Soret bands (in the range 380–410 nm) and in the Q-bands (520–560 nm range). On the basis of spectral and photophysical properties, as well as by comparison with corresponding literature data,1e,2,6,7 the emission is assigned to the lowest-lying singlet π–π* excited state, involving the corrole core, for all the compounds.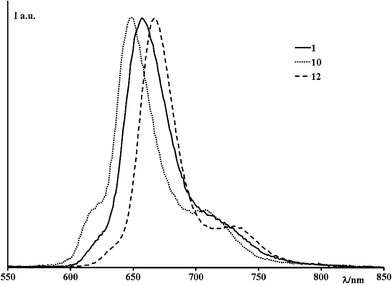 | ||
| Fig. 3 Normalized emission spectra of 1, 10 and 12 in dichloromethane solution. | ||
As indicated by the data in Table 1, the energy level of the emitting state is roughly constant for all the compounds, so indicating that the various pyrimidinyl substituents do not significantly affect corrole macrocycle π-orbitals. However, corroles 11 and 12 exhibit a slightly red-shifted emission, which parallels their absorption spectra red shift (see above) and can be similarly explained. On the contrary, significant differences are found as far as the fluorescence lifetimes and quantum yields of the various compounds are concerned (Table 1).
Fluorescence lifetimes and quantum yields of free-base corroles depend, together with other factors, on the grade of distortion of the corrole ring in the excited state in comparison with the ground state (it has been reported that distortion from planarity reduces radiative rate constants in comparison with radiationless rate constants, so decreasing fluorescence efficiency2b) and on the possible intervening of CT states, which contribute to the radiationless decay.2b,6,8 For example, the presence of nitrophenyl groups, which are good electron acceptors, as corrole substituents reduces significantly the luminescence lifetimes and quantum yields of the corrole core by the intervening of a low-lying CT state involving the nitrophenyl unit, which deactivates the corrole fluorescent state by oxidative electron transfer.2b We have proposed that the presence of a meso-dichloropyrimidinyl substituent leads to mixing between the strongly emissive π–π* corrole core excited state and a (less emissive or even radiationless) CT state in which the corrole core plays the role of the donor and the pyrimidinyl group can play the role of the acceptor.5 This suggestion was based on the fact that pyrimidinyl groups are known to behave as acceptors9 and corroles are known to be able to play the role of donors10 in CT states. Such an excited-state mixing could lead to reduction of the fluorescence efficiency, essentially due to increased radiationless rate constants. This effect appeared, however, to be deactivated when the chlorine atoms linked to the pyrimidinyl substituents were replaced by electron donor moieties such as phenoxy groups.6 The results obtained by the present study (data in Table 1), thanks to the availability of a large number of variously substituted compounds, allow to refine and correct the above mentioned statements, suggesting also new insights concerning the photophysical properties of pyrimidinyl-substituted corroles.
Analysis of data in Table 1 suggests that the studied compounds can be classified in subgroups, depending on their emission lifetimes and quantum yields:
– Compounds 1–7 (with the exception of compound 6) belong to one group (groupa), characterized by emission lifetimes in the narrow range 1.23–1.41 ns and by emission quantum yields in the range 0.020–0.038, with rate constants for radiationless decay, knr, in the range 6–8 × 108 s−1 (Table 1).
– Compounds 9 and 10 belong to a second group (groupb), with lifetimes and quantum yields close to 0.76 ns and 0.015, respectively, with knr to be around 1.3 × 109 s−1. Into this second group, corrole 6 can also be included (lifetime and quantum yield: 0.42 ns and 0.011, respectively, and knr is 2.4 × 109 s−1) (Table 1).
– Finally, a third group (groupc) includes corroles 8 and 11–14; these compounds exhibit emission lifetimes in the narrow range 4.11–4.81 ns and quantum yields larger than 0.06, up to 0.13. Radiationless decay rate constants for these species are in the range 1–2 × 108 s−1.
Apparently, the rate constant for radiative decay kr is roughly constant in the series of compounds (or anyway, there is no clear dependence on general structural features), ranging from 1.5 × 107 s−1 to 3.2 × 107 s−1 (Table 1).
The compounds belonging to the first group mentioned above are corroles bearing two meso-dichloropyrimidinyl substituents. In comparison with corroles without dichloropyrimidinyl substituents such as compound 8, where the electron-withdrawing effect of the pyrimidinyl substituent is largely balanced by the phenoxy group, and which exhibits a lifetime of 4.81 ns and a quantum yield of 0.132 in the same experimental conditions (Table 1), the corroles of groupa exhibit significantly shorter lifetimes and smaller quantum yields, essentially due to larger knr values.
Following our former hypothesis of the involvement of a low-lying CT state in deactivating the emissive state, it could be proposed that the reduction in emission efficiency for the compounds of groupa is due to the mixing of the emissive π–π* “pure” corrole-centered excited state with a doubly degenerated (since two dichloropyrimidinyls are equivalent) excited state having partial CT character, involving the two dichloropyrimidine substituents. As for the meso-dichloropyrimidinylcorroles having a single dichloropyrimidinyl group studied before,6 this mixing could translate into less efficient emissive properties. A representative of meso-pyrimidinyl-substituted corroles of this latter type (that is, carrying a single dichloropyrimidinyl unit) is compound A, whose emission lifetime and quantum yield are 2.4 ns and 0.066, respectively (knr = 3.8 × 108 s−1), both values intermediate between the values of 8 (without any dichloropyrimidinyl substituent) and those of the compounds in the groupa (carrying two dichloropyrimidinyl units) here studied, so in agreement with the “CT” interpretation. Compound 6, structurally similar to the other compounds of groupa, has a para-nitrophenyl substituent which further contributes to reduce its emission efficiency, in agreement with the findings of other nitrophenyl-containing corroles,2b so moving the properties of 6 into another group. This second group of compounds is groupb, to which also 9 and 10, A3-corroles bearing threedichloropyrimidinyl substituents, belong. The shorter lifetimes and smaller quantum yields of these compounds compared to those belonging to the groupa discussed above could be perfectly reasonable on considering that for 9 and 10three low-lying CT states can be mixed with the “pure” π–π* corrole-centered state, and the shortening of emission lifetimes and decreasing of emission quantum yields would therefore be more effective for the groupb compounds than for the groupa species (it should be clear now why 6, in which one CT state involving the nitrophenyl group is added to the doubly degenerated CT state involving two dichloropyrimidyl units, would be included in groupb, characterized by including compounds having three CT states mixed with the π–π* state).
The compounds in groupc seem to be structurally heterogeneous: the group in fact includes corroles with one (8), two (11 and 13) and even three (12 and 14) pyrimidinyl substituents. However, these species exhibit the longest lifetimes and the largest quantum yields of all the compounds studied here, in spite of the presence of multiple pyrimidinyl substituents. This circumstance cannot be a surprise as far as compounds 8, 13 and 14 are concerned, as it has already been proposed that phenoxy substituents on the pyrimidinyl groups destabilize the CT states, making the effect of the presence of pyrimidinyl electron acceptor substituents ineffective in modifying the corrole photophysical properties.6 However, our results show that also corroles carrying only phenyl-substituted pyrimidinyl groups (e.g., compounds 11 and 12), expected to be more neutral from an electronic viewpoint, roughly behave as species with phenoxy-substituted pyrimidinyl moieties.
Whereas the hypothesis of the CT state effect could fit the experimental data of the compounds here studied to some extent, another hypothesis has to be considered. It has recently been proposed that the presence of halogens as substituents at meso-phenyl groups of corroles can introduce heavy-atom effects on their photophysical properties, so accelerating intersystem crossing and reducing fluorescence lifetimes and quantum yields.11 This effect is well known in the spectroscopy of porphyrins.12 The classification of the compounds here investigated within groupsa, b and c as a function of their photophysical properties, in casu their knr values (calculated by using fluorescence lifetimes and quantum yields) reported in Table 1, also fits with the number of chlorine atoms linked in the ortho-position to the meso-pyrimidinyl substituents. The ortho-position indeed leads the substituents of the pyrimidinyl groups close to the corrole ring, enabling perturbations of the corrole-based excited state dynamics (e.g. heavy-atom enhanced intersystem crossing) are possible. In fact, compound 2 contains an extra chlorine atom in the para-position of the phenyl substituent, but this chlorine atom is remote from the corrole core and apparently does not have a noticeable effect on knr. The knr value is indeed the sum of two rate constants, due to two different radiationless processes, kIC, internal conversion leading to the ground state population (unaffected by heavy-atom effects) and kISC, intersystem crossing to the π–π* triplet state, which depends on spin–orbit coupling and therefore largely affected by the effective presence of heavy atoms. As a consequence, the results reported here can be attributed to the presence of a specific number of chlorine atoms in ortho positions to meso-aryl substituents, with compounds of the above mentioned groupa having four chlorine atoms, compounds in groupb having six chlorine atoms, and compounds in groupc being free of chlorine atoms (note that the reference compound A has two chlorine atoms).
Within this latter hypothesis, the only compound of the investigated species in which a genuine CT state would be present is 6, in which the CT state involves the nitrophenyl group. Indeed for 6, assuming oxidative electron transfer takes place, as reported for a similar corroles and porphyrins,2a,13 the electron transfer quenching rate constant kel can be estimated, considering that the singlet state decay rate of 6 is 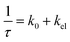 , where k0 is the reciprocal fluorescence lifetime for parent unsubstituted compound 1. The calculated value of kel is as high as 1.58 × 109 s−1. Interestingly, subtraction of this kel value from the measured knr value (listed in Table 1) gives 7.75 × 108 s−1 for the radiationless decay processes rate constant due to the sum of intersystem crossing and internal conversion processes, a figure in line with those of the other compounds of groupa.
, where k0 is the reciprocal fluorescence lifetime for parent unsubstituted compound 1. The calculated value of kel is as high as 1.58 × 109 s−1. Interestingly, subtraction of this kel value from the measured knr value (listed in Table 1) gives 7.75 × 108 s−1 for the radiationless decay processes rate constant due to the sum of intersystem crossing and internal conversion processes, a figure in line with those of the other compounds of groupa.
The literature data for compound B6 (Fig. 1) favour the enhanced intersystem crossing hypothesis. Actually, fluorescence lifetime and quantum yield for B in air-equilibrated dichloromethane solution at room temperature are 0.8 ns and 0.020, respectively (see Table 1). These values yield a radiationless decay rate constant knr equivalent to 1.23 × 109 s−1. By taking into account the “CT” hypothesis, compound B should exhibit properties similar to that of A, containing a single dichloropyrimidinylmeso-substituent, whereas according to the “intersystem crossing” hypothesis it should belong to groupb, as it contains six chlorine atoms. Since the calculated knr of B is close to those of 9 and 10 (see Table 1), the compounds identifying groupb, it can be tentatively suggested that the determining factor leading to reduced fluorescence lifetime and quantum yield in the present corrole compounds is increased spin–orbit coupling induced by the presence of heavy atoms such as chlorine. The nature of the aryl group itself (pyrimidinyl/phenyl) and contributions originating from closely-lying CT states (if any) would play only minor roles.
To further examine the hypothesis based on increased spin–orbit coupling due to the presence of chlorine atoms in the vicinity of the corrole macrocycle to rationalize the luminescence data, it has to be noted that, although the spin–orbit coupling constant value for a chlorine atom is not that high (ζ = 587 cm−1), the rate constant depends on the spin–orbit coupling constant squared. Increase in chlorine number from two to six may well provide substantial changes in the photophysical properties. A decrease in the fluorescence quantum yield Φf and enhancement of the intersystem crossing rate constant kISC should be observed due to heavy atom effects, whereas the internal conversion rate kIC should not be influenced.14 Therefore, the changes in the measured radiationless deactivation rate knr (i.e., the sum of kISC and kIC, as previously stated) can be directly related to changes in the intersystem crossing rate kISC, i.e. Δknr = ΔkISC. Fig. 4 represents the dependence of the radiationless deactivation rate knr in the studied corroles on the sum of the spin–orbit coupling constant squared contributed by the chlorine atoms. A good linear dependence is observed indicating the progressive rise of the intersystem crossing rate with increasing the number of chlorine atoms. This dependence is a clear signature of the heavy atom effect on the photophysical properties of 4,6-dichloropyrimidinyl-substituted corroles. The dependence of the fluorescence quantum yield Φf on the sum of spin–orbit coupling constants squared of the chlorine atoms is shown in Fig. 5. The dependence has the same trend of progressive Φf value changes with increase in substituted chlorine atom number. In this case the dependence is a decreasing function since the increasing kIC value is in the denominator in the relevant equation 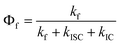 . The fluorescence quantum yields Φf have distinctly different values for the mono-, bis- and tris(4,6-dichloroaryl)-substituted corroles. However, one should note that the scatter of Φf values for disubstituted corroles 1–7 is larger, indicating that the fluorescence quantum yield reflects the individuality of the B-substituents to a larger extent than the knr value does. But if we analyse separately the series with the same type of substituents (i.e.A, 7, 9 and 1, 10) separately, the slope of Φf dependence in both cases remains about the same. This fact further supports the hypothesis that the change in the intersystem crossing rate is probably the factor determining the observed trend of fluorescence quantum yield with increase in the number of chlorine atoms.
. The fluorescence quantum yields Φf have distinctly different values for the mono-, bis- and tris(4,6-dichloroaryl)-substituted corroles. However, one should note that the scatter of Φf values for disubstituted corroles 1–7 is larger, indicating that the fluorescence quantum yield reflects the individuality of the B-substituents to a larger extent than the knr value does. But if we analyse separately the series with the same type of substituents (i.e.A, 7, 9 and 1, 10) separately, the slope of Φf dependence in both cases remains about the same. This fact further supports the hypothesis that the change in the intersystem crossing rate is probably the factor determining the observed trend of fluorescence quantum yield with increase in the number of chlorine atoms.
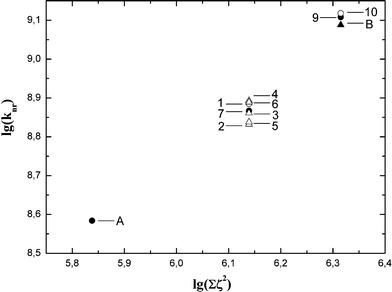 | ||
| Fig. 4 Dependence of knr on the sum of chlorine spin orbit coupling constant squared (logarithmic scale). The value of knr of 6 used here is the one obtained by subtracting the kel value from total knr measured (see text). | ||
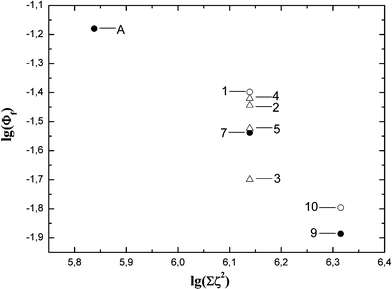 | ||
| Fig. 5 Dependence of emission quantum yields on the sum of chlorine spin orbit coupling constant squared (logarithmic scale). | ||
To help in the discrimination between the “CT” and “enhanced intersystem crossing” mechanisms, we performed experiments at 77 K in toluene on selected compounds: in fact, in these conditions the CT process is expected to be disfavored, and this could lead to an increased emission lifetime. However, the 77 K luminescence lifetimes of 1, 9, and A (1.9 ns, 0.8 ns, and 2.6 ns, respectively) do not change significantly from their room temperature values (Table 1). Whereas this cannot be taken as a decisive argument for definitely excluding a CT mechanism, surely it further supports the “enhanced intersystem crossing” explanation. It can be also noted that the emission lifetime of 6, where a CT state involving the nitrophenyl group subunit is surely active, increases to 2.2 ns at 77 K.
The luminescence properties of the compounds 8 and 11–14 warrant some additional comments. These corroles, in particular 11–14, have bulky groups at the ortho,ortho′- positions of the meso-pyrimidinyl substituents. Bulky groups may limit free rotation of the corrole substituents and, more importantly, may promote nonplanar distortions of the corrole macrocycle. The presence of nonplanar distortions in tetrapyrrolic compounds is known to lead to strong enhancement of the nonradiative deactivation rate and fluorescence quenching.2b,15 However the data in Table 1 indicate that the knr values in this series of compounds do not follow any trend based on the presence of bulky substituents (for example, compound 14 has more bulky groups than 13, but their knr values are almost identical one another). The fluorescence lifetimes and yields are close to those measured for mesityl- and pentafluorophenyl-substituted corroles, which are known to produce no noticeable macrocycle distortions.7a Hence, whereas free rotation of the pyrimidinyl substituents is most likely limited when bulky groups are present in the studied compounds, these steric interactions are apparently not strong enough to promote nonplanar distortions of the corrole macrocycle. This is an important observation, since compounds such as 13 and 14 can be used as a core in the design of corrole-centered multichromophoric (dendritic) systems, without affecting the photophysical properties of the corrole macrocycle.
Finally, it could also be noted that, according to the “intersystem crossing hypothesis”, the kISC in compounds such as 9 and 10 should dominate the radiationless decay. In fact, assuming knr in non-chlorine containing compounds is mainly due to kIC and considering the acceleration of knr in 9 and 10 as exclusively due to increased kISC, kISC in the latter species should be several times larger than kIC. This would suggest that the triplet π–π* state in compounds like 9 and 10 is produced with a high efficiency (about 70–80%).16 Because triplet state formation is a prerequisite for the reported intriguing properties of corroles in photodynamic therapy,17 our results appear to be quite interesting for future design of corrole compounds with potential therapeutical applications as well as for other applications based on triplet excited states.
Conclusions
The luminescence properties of a series of corroles containing multiple pyrimidinyl substituents have been studied. In particular, nine corroles containing two pyrimidinyl groups, four corroles containing three pyrimidinyl groups and one corrole carrying a single pyrimidinyl substituent have been investigated, and their properties have been compared with a couple of model species. The results indicate that the energy of the emissive π–π* corrole-core based state is not perturbed significantly by the various substituents, whereas the emission lifetimes and quantum yields depend on the number of appended meso-dichloropyrimidinyl substituents. Both emission lifetime and quantum yield decrease with increasing the number of meso-dichloropyrimidinyl substituents, whereas pyrimidinyl substituents which do not carry further electron withdrawing groups such as the chlorine substituents do not affect the corrole emission properties. These results can be rationalized by two hypotheses: (i) on assuming that the presence of meso-dichloropyrimidinyl substituents introduces low-lying CT states which mix with the corrole π–π* emissive level, so reducing emission efficiency; (ii) on considering that the ortho,ortho′-chlorine groups of the meso-pyrimidinyl substituents can increase the intersystem crossing rate constant of the corrole-based fluorescent state via the heavy-atom effect. The analysis of the results for the studied compounds together with elaboration of photophysical data for recently studied species tend to suggest that the hypothesis (ii) is the most likely one, although contribution from CT states cannot be definitely excluded. To this aim, comparison of the photophysical properties of the present compounds with those of analogous Br-substituted compounds, whose preparation is in progress in our laboratories, could offer a decisive solution.The results are of particular interest for the design of corrole compounds with highly efficient triplet formation and for the preparation of multicomponent systems containing photo-active corrole subunits and pyrimidinyl spacers, which is in progress in our laboratories.
Experimental section
The compounds investigated have been synthesized as formerly reported.5,6 For the photophysical experiments, solvents of the higher spectroscopic grade were used. Extreme care was taken to eliminate any traces of acids or bases in the solutions. Absorption spectra were performed with a Jasco V560 spectrophotometer; luminescence spectra were performed with a Horiba Jobin Yvon Spex Fluoromax P fluorimeter, equipped with a red sensitive Hamamatsu photomultiplier. The spectra were corrected with software purchased by the manufacturer. Luminescence lifetimes have been measured with an Edinburgh OB900 time-correlated single-photon-counting spectrometer, using a Hamamatsu PL2 laser diode as excitation pulse (time pulse, 59 ps at 408 nm). Luminescence quantum yields have been calculated by using the optically dilute method18 and using [Ru(bpy)3]2+ (bpy = 2,2′-bipyridine) in air-equilibrated aqueous solution as standard (Φ = 0.028).19Experimental uncertainties are as follows: absorption maxima: 2 nm; emission maxima: 4 nm; molar absorption: 10%; emission lifetimes: 10%; emission quantum yields: 10%.
Acknowledgements
The authors thank the IWT (Institute for the Promotion of Innovation through Science and Technology in Flanders) for a doctoral fellowship to T. H. Ngo, the FWO (Fund for Scientific Research – Flanders) for financial support and a postdoctoral fellowship to W. Maes, and the KU Leuven and the Ministerie voor Wetenschapsbeleid for continuing financial support. The University of Messina (Progetti di Ricerca di Ateneo to S.C. and Fondi per Giovani Ricercatori to F.N.) and MIUR (PRIN 2008 projects) are also acknowledged for financial support. M. Kruk thanks the State Program of Scientific Research of the Republic of Belarus for financial support.References
- (a) Z. Gross, N. Galili and I. Saltsman, The First Direct Synthesis of Corroles from Pyrrole, Angew. Chem., Int. Ed., 1999, 38, 1427–1429 CrossRef CAS; (b) R. Paolesse, L. Jaquinod, D. J. Nurco, S. Mini, F. Sagone, T. Boschi and K. M. Smith, 5,10,15-Triphenylcorrole: a product from a modified Rothemund reaction, Chem. Commun., 1999, 1307–1308 RSC; (c) J.-M. Barbe, G. Canard, S. Brandès and R. Guilard, Selective Chemisorption of Carbon Monoxide by Organic–Inorganic Hybrid Materials Incorporating Cobalt(III) Corroles as Sensing Components, Chem.–Eur. J., 2007, 13, 2118–2129 CrossRef CAS; (d) I. Aviv and Z. Gross, Corrole-based applications, Chem. Commun., 2007, 1987–1999 RSC; (e) L. Flamigni and D. T. Gryko, Photoactive corrole-based arrays, Chem. Soc. Rev., 2009, 38, 1635–1646 RSC; (f) I. Aviv-Harel and Z. Gross, Aura of Corroles, Chem.–Eur. J., 2009, 15, 8382–8394 CrossRef CAS.
- (a) R. Paolesse, F. Sagone, A. Macagnano, T. Boschi, L. Prodi, M. Montalti, N. Zaccheroni, F. Bolletta and K. M. Smith, Photophysical Behaviour of Corrole and its Symmetrical and Unsymmetrical Dyads, J. Porphyrins Phthalocyanines, 1999, 3, 364–370 CrossRef CAS; (b) R. Paolesse, A. Marini, S. Nardis, A. Froiio, F. Mandoj, D. J. Nurco, L. Prodi, M. Montalti and K. M. Smith, Novel routes to substituted 5,10,15-triarylcorroles, J. Porphyrins Phthalocyanines, 2003, 7, 25–36 CrossRef CAS.
- (a) D. T. Gryko, Recent Advances in the Synthesis of Corroles and Core-Modified Corroles, Eur. J. Org. Chem., 2002, 1735–1743 CrossRef CAS; (b) D. T. Gryko, J. P. Fox and P. Goldberg, Recent advances in the chemistry of corroles and core-modified corroles, J. Porphyrins Phthalocyanines, 2004, 8, 1091–1105 CrossRef CAS; (c) A. Ghosh, A Perspective of One-Pot Pyrrole–Aldehyde Condensations as Versatile Self-Assembly Processes, Angew. Chem., Int. Ed., 2004, 43, 1918–1931 CrossRef CAS; (d) R. Paolesse, Corrole: The Little Big Porphyrinoid, Synlett, 2008, 2215–2230 CrossRef CAS; (e) D. T. Gryko, Adventures in the synthesis of meso-substituted corroles, J. Porphyrins Phthalocyanines, 2008, 12, 906–917 CrossRef CAS.
- (a) M. Tasior, D. T. Gryko, M. Cembor, J. S. Jaworski, B. Ventura and L. Flamigni, Photoinduced energy and electron transfer in 1,8-naphthalimide–corrole dyads, New J. Chem., 2007, 31, 247–259 RSC; (b) L. Flamigni, B. Ventura, M. Tasior, T. Becherer, H. Langhals and D. T. Gryko, New and Efficient Arrays for Photoinduced Charge Separation Based on Perylene Bisimide and Corroles, Chem.–Eur. J., 2008, 14, 169–183 CrossRef CAS; (c) M. Tasior, D. T. Gryko, J. Shen, K. M. Kadish, T. Becherer, H. Langhals, B. Ventura and L. Flamigni, Energy- and Electron-Transfer Processes in Corrole–Perylenebisimide–Triphenylamine Array, J. Phys. Chem. C, 2008, 112, 19699–19709 CrossRef CAS; (d) A. Ghosh, T. Wondimagegn and A. B. J. Parusel, Do Nonplanar Porphyrins Have Red-Shifted Electronic Spectra? A DFT/SCI Study and Reinvestigation of a Recent Proposal, J. Am. Chem. Soc., 2000, 122, 6371–6374 CrossRef CAS; (e) M. Tasior, D. T. Gryko, D. J. Pielacinska, A. Zanelli and L. Flamigni, Trans-A2B-corroles Bearing a Coumarin Moiety - From Synthesis to Photophysics, Chem.–Asian J., 2010, 5, 130–140 CrossRef CAS; (f) L. Shi, H.-Y. Liu, K.-M. Peng, X.-L. Wang, L.-L. You, J. Lu, L. Zhang, H. Wang, L.-N. Ji and H.-F. Jiang, Synthesis of phenothiazine-corrole dyads: the enhanced DNA photocleavage properties, Tetrahedron Lett., 2010, 51, 3439–3442 CrossRef CAS.
- (a) W. Maes, T. H. Ngo, J. Vanderhaeghen and W. Dehaen, meso-Pyrimidinyl-Substituted A2B-Corroles, Org. Lett., 2007, 9, 3165–3168 CrossRef CAS; (b) T. H. Ngo, W. Van Rossom, W. Dehaen and W. Maes, Reductive demetallation of Cu-corroles–a new protective strategy towards functional free-base corroles, Org. Biomol. Chem., 2009, 7, 439–443 RSC; (c) T. H. Ngo, F. Nastasi, F. Puntoriero, S. Campagna, W. Dehaen and W. Maes, meso-Pyrimidinyl-Substituted A2B- and A3-Corroles, J. Org. Chem., 2010, 75, 2127–2130 CrossRef CAS.
- T. H. Ngo, F. Puntoriero, F. Nastasi, K. Robeyns, L. Van Meervelt, S. Campagna, W. Dehaen and W. Maes, Synthetic, Structural, and Photophysical Exploration of meso-Pyrimidinyl-Substituted AB2-Corroles, Chem. Eur. J., 2010, 16, 5691–5705 CAS.
- (a) B. Ventura, A. Degli Esposti, B. Koszarna, D. T. Gryko and L. Flamigni, Photophysical characterization of free-base corroles, promising chromophores for light energy conversion and singlet oxygen generation, New J. Chem., 2005, 29, 1559–1566 RSC; (b) B. Koszarna and D. T. Gryko, Meso–meso linked corroles, Chem. Commun., 2007, 2994–2996 RSC.
- T. Ding, E. A. Aleman, D. A. Modarelli and C. J. Ziegler, Photophysical Properties of a Series of Free-Base Corroles, J. Phys. Chem. A, 2005, 109, 7411–7417 CrossRef CAS.
- R. Passalacqua, F. Loiseau, S. Campagna, Y. Q. Fang and G. S. Hanan, In Search of Ruthenium(II) Complexes Based on Tridentate Polypyridine Ligands that Feature Long-lived Room-Temperature Luminescence: The Multichromophore Approach, Angew. Chem., Int. Ed., 2003, 42, 1608–1611 CrossRef CAS.
- See, for example: F. D'Souza, R. Chitta, K. Ohkubo, M. Tasior, N. K. Subbaiyan, M. E. Zandler, M. K. Rogacki, D. T. Gryko and S. Fukuzumi, Corrole–Fullerene Dyads: Formation of Long-Lived Charge-Separated States in Nonpolar Solvents, J. Am. Chem. Soc., 2008, 130, 14263–14272 Search PubMed.
- (a) L. Shi, H.-Y. Liu, H. Shen, J. Hu, G.-L. Zhang, H. Wang, L.-N. Ji, C.-K. Chang and H.-F. Jiang, Fluorescence properties of halogenated mono-hydroxyl corroles: the heavy-atom effects, J. Porphyrins Phthalocyanines, 2009, 13, 1221–1226 CrossRef CAS; (b) L. L. You, H. Shen, L. Shi, G. L. Zhang, H. Y. Liu, H. Wang and L. N. Ji, Photophysical properties of the corrole photosensitizers, Sci. China, Ser. G: Phys., Mech. Astron., 2010, 53, 1491–1496 Search PubMed.
- (a) E. G. Azenha, A. C. Serra, M. Pinero, M. M. Pereire, J. Seixas de Melo, L. G Arnaut, S. J. Formosinho, A. M.d’ and A. Rocha Gonsalves, Heavy-atom effects on metalloporphyrins and polyhalogenated porphyrins, Chem. Phys., 2002, 280, 177–190 CrossRef CAS; (b) G. D. Egorova, V. N. Knyukshto, K. N. Solovyov and M. P. Tsvirko, Intramolecular spin-orbital perturbations in ortho- and meta-halo derivatives of tetraphenylporphine, Opt. Spectrosc., 1980, 48, 1101–1109 CAS; (c) K. N. Solovyov and E. A. Borisevich, Intramolecular heavy-atom effect in the photophysics of organic molecules, Prog. Phys., 2005, 175, 247–270 Search PubMed.
- (a) R. A. Binstead, M. J. Crossley and N. S. Hush, Modulation of valence orbital levels of metalloporphyrins by β-substitution: evidence from spectroscopic and electrochemical studies of 2-substituted metallo-5,10,15,20-tetraphenylporphyrins, Inorg. Chem., 1991, 30, 1259–1264 CrossRef CAS; (b) S. Dahal and V. Kroshnan, Excited singlet state intramolecular charge transfer in di and trinitrotetraphenylporphyrins, Chem. Phys. Lett., 1997, 274, 390–395 CrossRef CAS; (c) V. N. Knyukshto, E. Zenkevich, E. Sagun, A. Shulga and S. Bachilo, Pathways for photoinduced electron transfer in meso-nitro-phenyl-octaethylporphyrins and their chemical dimers, Chem. Phys. Lett., 1999, 304, 155–166 CrossRef CAS; (d) V. A. Walters, J. C. de Paula, B. Jackson, C. Ntaitis, K. Hall, J. Lind, K. Cardozo, K. Chandran, D. Raible and C. M. Phollips, Electronic Structure of Triplet States of Zinc(II) Tetraphenylporphyrins, J. Phys. Chem., 1995, 99, 1166–1171 CrossRef CAS.
- S. P. McGlynn, T. Azumi, M. Kinoshita, Molecular Spectroscopy of the triplet state, Prentice Hall, Inc., Englewood Cliffs, New Jersey, 1969 Search PubMed.
- B. Röder, M. Büchner, I. Rückmann and M. O. Senge, Correlation of photophysical parameters with macrocycle distortion in porphyrins with graded degree of saddle distortion, Photochem. Photobiol. Sci., 2010, 9, 1152–1158 RSC.
- In a first approximation, kISC in 9 should be larger than 1 × 109 s−1 (kISC = knr − kIC, where knr is the experimentally calculated radiationless rate constant, 12.8 × 108 s−1, and kIC could be approximated to the experimental radiationless decay of any of the compounds 11–14, that is about 2.5 × 108 s−1). As the emission quantum yield of 9 is 0.013, more than 98% of the excited state deactivates by radiationless decay, and more than 80% of the radiationless decay is due to intersystem crossing. With this figure, the triplet π–π* state is produced with an efficiency of about 80% from the singlet state. Additional indirect evidence of the high efficiency of triplet state population and enhancement upon introducing of halide atoms into aryl substituents can be derived from comparison of our results with literature data, where it was found that triphenylcorrole triplet state quantum yield is only 0.51, while pentafluorophenylcorrole has triplet state quantum yield as high as 0.71 (see ref. 7a).
- C. K. Chang, P.-W. Kong, H.-Y. Liu, L. L. Yeung, H.-K. Koon and N.-K. Mak, Synthesis and photodynamic activities of modified corrole derivatives on nasopharyngeal carcinoma cells, Proc. SPIE, 2006, 6139, 268–278.
- G. A. Crosby and J. N. Demas, Measurement of photoluminescence quantum yields. Review, J. Phys. Chem., 1971, 75, 991–1024 CrossRef.
- N. Nakamaru, Synthesis, Luminescence Quantum Yields, and Lifetimes of Trischelated Ruthenium(II) Mixed-ligand Complexes Including 3,3′-Dimethyl-2,2′-bipyridyl, Bull. Chem. Soc. Jpn., 1982, 55, 2697–2705 CAS.
| This journal is © The Royal Society of Chemistry and Owner Societies 2011 |