Photophysics and reductive quenching reactivity of gadusol in solution
Ernesto Maximiliano
Arbeloa
ab,
Sonia Graciela
Bertolotti
bc and
María Sandra
Churio
*ab
aDepartamento de Química, Facultad de Ciencias Exactas y Naturales, Universidad Nacional de Mar del Plata, Funes 3350, B7602AYL Mar del Plata, Argentina. E-mail: schurio@mdp.edu.ar; Fax: +54 223 4753150; Tel: +54 223 4756167
bConsejo Nacional de Investigaciones Científicas y Técnicas (CONICET), Argentina
cDepartamento de Química, Facultad de Ciencias Exactas, Fisico-Químicas y Naturales, Universidad Nacional de Río Cuarto, Campus Universitario. km 601, X5804ALH Río Cuarto, Argentina
First published on 12th November 2010
Abstract
The photostability and photophysics of gadusol in aqueous solution has been studied. The photodecomposition quantum yields (ca. 4 × 10−2 and 1 × 10−4 at acidic and neutral pH, respectively) confirm the high photostability of the metabolite, independently of the presence of oxygen, under physiological conditions. The nature of the electronic transition of gadusol has been assigned as π → π* on the basis of the solvatochromic shifts of the UV absorption spectrum and the time-dependent density functional theory calculation of the vertical transition energies. The results from the photoacoustic calorimetry point to the rapid non-radiative decay as the dominant relaxation pathway of the excited species at pH 7, which is consistent with the proposed UV-sunscreening role of the molecule in the early atmosphere. Laser flash photolysis experiments probed that the ground state of the enolate form (gadusolate) undergoes electron transfer reactions with some triplet sensitizers in water or methanol solution. A rate constant of 2 × 108 M−1s−1 has been determined for the quenching of rose bengal triplet state in water at pH 7. This reductive quenching reactivity may be considered as one of the underlying mechanisms that support the antioxidant capacity of gadusol in biological environments.
Introduction
The natural product (3,5,6-trihydroxy-5-hydroxymethyl-2-methoxycyclohex-2-en-1-one), known as gadusol, is structurally related to the mycosporine-like amino acids (MAAs), a family of substances occurring in a wide variety of marine and terrestrial organisms from tropical to Antarctic latitudes.1,2 The presence of this metabolite in the ovaries and developing larvae of marine fishes,3–6 sea urchin eggs,7 cysts and nauplii of the brine shrimp Artemia,8 and sponges,9 has led to the invocation of a role in reproduction. However gadusols have also been found in association with soluble proteins in fish lenses and probably in the outer dorsal skin layers of some fish species.10,11Biochemical transformations linking gadusol and MAAs have been suggested.12 In fact biosynthesis and/or trophic or symbiotic transference have been addressed as the origins of MAAs in the organisms. Some experimental evidence supports de novo synthesis of MAAS through a branch of the shikimate pathway.13,14 Both gadusol and the simpler structure deoxygadusol (3,5-dihydroxy-5-hydroxymethyl-2-methoxycyclohex-2-en-1-one) are presumed to be immediate precursors and the intermediates between MAAs and the shikimate biosynthetic route.2 Moreover, deoxygadusol has been prepared by retrobiosynthesis via bacterial conversion from the MAAs shinorine and porphyra-334.15,16
Most of the interest concentrated on MAAs during the last decades is related to their inferred UV-photoprotective role in living organisms.2,10,17,18 While UV absorption maxima of MAAs range between 310 and 360 nm, gadusols absorb strongly towards the UVB and UVC zones with pH-dependent distinctive maxima: λmax(H2O, pH < 2)/nm 269 (ε/dm3 mol−1 cm−1 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400) and 296 (21800) at pH > 7,5 as expected from the shift in the acid–base equilibrium of the enol tautomer of a 1,3-diketone with a pKa = 4.2.†4,5,19
400) and 296 (21800) at pH > 7,5 as expected from the shift in the acid–base equilibrium of the enol tautomer of a 1,3-diketone with a pKa = 4.2.†4,5,19
The photochemistry and the photophysics of the MAAs are relevant aspects to understanding the mechanisms of their photoprotective activity. The photostability of the imino-MAAs shinorine, porphyra-334 and palythine has been assessed in vitro.18,20–22 The low photodecomposition quantum yields together with the negligible fluorescence and the non-radiative deactivation, as the main relaxation pathway of the excited states, support the proposed physiological function in UV-photoprotection.18,20,21
Particularly, the nature of the excited states of MAAs and related gadusols has not been evaluated before. In addition, except for the recent study on the mycosporine-glutaminol-glucoside,23 neither the photochemical properties of mycosporines and oxo-MAAs nor the photoinduced processes on gadusols in solution have been characterized.
The broad presence of MAAs and related gadusols in marine organisms has been connected to a potential antioxidant protection. In fact, oxo-MAAs such as mycosporine glycine and mycosporine-taurine have been found to possess a moderate antioxidant capacity, whereas imino-MAAs such as porphyra-334 and shinorine, seem to be oxidatively robust.24,25 The antioxidant capacity of deoxygadusol has been evaluated through the phosphatidylcholine peroxidation inhibition-assay, yielding higher activity than mycosporine-glycine.26 Besides, a significant efficiency for the quenching of singlet oxygen has been determined for deoxygadusol in a solvent mixture of methanol and chloroform.27
Other assays have demonstrated that redox properties of gadusol are similar to those of ascorbic acid.2,5 Recently, the oxygen radical absorbance capacity (ORAC) and the 2,2′-azinobis(3-ethylbenzothiazoline-6-sulfonic acid) diammonium salt (ABTS) tests have confirmed that gadusol is comparable to ascorbic acid towards reductive reactions of radicals but has stronger ability to break chain reactions carried by peroxyl radicals.6
Some interesting speculations have been raised about the role played by oxo-MAAs and gadusols in the evolution of phototrophic life on the early Earth. It has been proposed that the requirement for UV protection by living organisms that developed in the absence of atmospheric O2 may have been fulfilled by gadusols, which evolved prior to MAAs and may have served originally as UVB/C screens.2,28 Concomitantly, the strong antioxidant properties of gadusols would have protected early cyanobacteria against oxidative damage at the intracellular sites of oxygenic photosynthesis.29 Later in evolution, amine condensation with deoxygadusol provided MAAs with strong UVB- and UVA-absorbing characteristics as rising atmospheric oxygen levels increased the need for protection against UVA and photooxidative stress.30
In this context, a full characterization of the photochemical and photophysical properties of gadusols in solution may contribute to validate the former theories. Besides, the role of the metabolites in the mitigation of the oxidative stress conditions in vivo is deeply entailed with the facility of energy and electron transfer in photosensitized processes, which needs to be examined.
In this paper we report on the photostability and absorption properties of gadusol and its anion gadusolate, and on the nature of both the electronic transitions and the relaxation pathways of their excited states. We also analyze the quenching reactivity of gadusolate with triplet sensitizers under physiological pH conditions and the potential contribution of this mechanism to the antioxidant capacity of the metabolite.
Results
Experimental and calculated UV absorption
The UV absorption spectrum of gadusol was examined in different solvent media. In aqueous solution, the reversible bathochromic shift of the maximum from 268 nm (pH 2.5) to 296 nm (pH ≥ 7) was observed together with an increase of the intensity (Fig. 1). High-performance liquid chromatography (HPLC) analysis on samples at each pH condition revealed a unique peak with distinctive retention times of ca. 6 and 4 min, at pH 2.5 and 7, respectively.6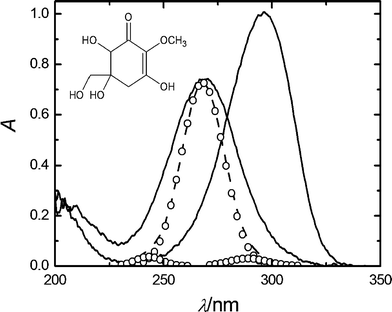 | ||
| Fig. 1 UV absorption spectra of 6 × 10−5 M gadusol in water at pH 2.5 (full line, λmax = 268 nm) and pH 7 (full line, λmax = 296 nm). Simulated absorption spectra (dotted line), built from the sum of the Gaussian functions (open circles) centred respectively on the calculated transitions (see text). The inset shows the chemical structure of gadusol, the major species in the acidic solution.4,5 | ||
The absorption spectra in acetonitrile–water mixtures shift to longer wavelengths on going from 100% acetonitrile (λmax = 263 nm) to 80% v/v (λmax = 268 nm). Besides, the absorption peaks in various alcoholic solvents appear around 269 nm. The data for the maximal wavelengths for these and other organic solvents were correlated with the normalized parameter of the solvent polarity ETN.31,32 A general trend of bathochromic shifts with the increasing polarity of the medium is verified (Fig. 2).
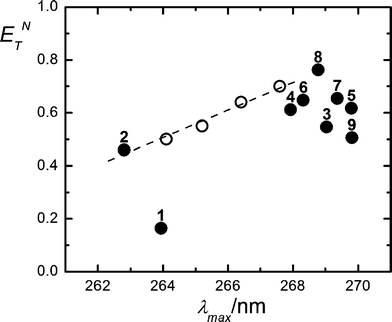 | ||
| Fig. 2 E T N parameter vs. maximum absorbance wavelengths for gadusol. Full symbols denote neat solvents: 1,4-dioxane (1); acetonitrile (2); 2-propanol (3); propanoic acid (4); 1-propanol (5); acetic acid (6); ethanol (7); methanol (8) and 2-butanol (9). Open symbols stand for acetonitrile–water mixtures (100, 98, 95, 90 and 80% v/v from left to right). ETN values were extracted from references 31 (neat solvents) and 32 (solvent mixtures). | ||
The correlation of the solvatochromic shifts by a multiparametric approach based on the Kamlet–Taft equation led to the results shown in Fig. 3. The linear fit of the data is described by λ = 266.44 − 0.079π* + 0.041α + 0.044β, with a correlation R factor of 0.9913.
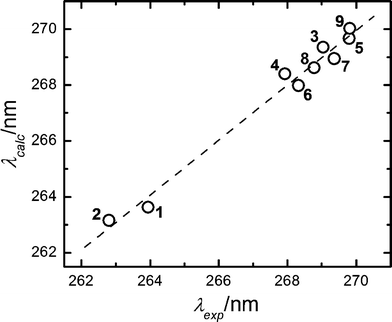 | ||
| Fig. 3 Calculated vs. experimental maximum absorbance wavelengths for gadusol in 1,4-dioxane (1); acetonitrile (2); 2-propanol (3); propanoic acid (4); 1-propanol (5); acetic acid (6); ethanol (7)); methanol (8) and 2-butanol (9). Calculated maximal absorption wavelengths follow the expression: λ = 266.44 − 0.079π* + 0.041α + 0.044β, according to the fit of the data to the Kamlet–Taft equation with R = 0.9913. | ||
A time-dependent density functional theory (TD-DFT) calculation of the Franck–Condon vertical transition energies for the enol form of gadusol in water was carried out in order to reproduce the experimental absorption spectrum in acidic solution. Various atomic basis sets were tested, which led to three transitions with similar characteristics. The largest oscillator strength belongs to the transition centred between 264 and 265 nm (blue-shifted ca. 3 nm from the experimental value), the other two weaker transitions lay around 240 and 285 nm. Fig. 1 qualitatively compares the experimental absorption spectrum with that one simulated by addition of a Gaussian function centred at each of the calculated transition wavelengths. The theoretical spectrum has been shifted 3 nm to the right for comparative purposes. In this case, the basis function that yielded the best fit to the experimental band (6-311++G*(Pauling)) was considered in order to calculate the set of molecular orbitals (MOs). The MOs involved in the three transitions are shown in Fig. 4. The most intense transition connects the orbitals designed as HOMO and LUMO, whereas weaker bands arise respectively from the transitions HOMO−2→LUMO (ca. 240 nm) and HOMO−1→LUMO (ca. 286 nm).
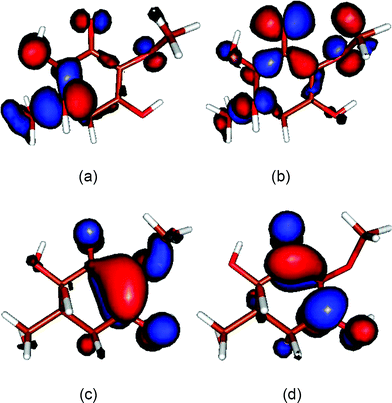 | ||
| Fig. 4 Molecular orbitals obtained for the enol form of gadusol in aqueous solution at the DFT B3LYP/6-311++G*(Pauling) theory level. (a) HOMO−2 (b) HOMO−1 (c) HOMO (d) LUMO. | ||
Emission and photostability
The fluorescence properties of gadusol in water were assessed under neutral and acid pH. No emission was observed from gadusol by excitation at 268 nm under acid pH or from gadusolate at 296 nm (neutral conditions), other than that assigned to the solvent medium according to the blank experiments.The photostabilities of gadusol and gadusolate were evaluated in the presence or absence of oxygen under steady monochromatic irradiation (Fig. 5). No new bands appeared in the 200–900 nm range of the absorption spectra of the irradiated samples. The simultaneous HPLC analyses of the irradiated mixtures at regular intervals of time were consistent with the absence of products absorbing in the spectral region of gadusols.
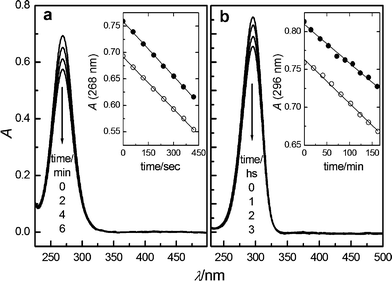 | ||
| Fig. 5 UV absorption spectra of (a) gadusol (pH 2.5) and (b) gadusolate (pH 7) at selected times, indicated by the arrows, during the stationary photolysis of initial 5 × 10−5 M Ar-bubbled water solutions. Insets: absorbance at λmaxvs. irradiation time for the photolysis under air (●) and Ar (○) atmospheres. | ||
The slopes of the linear fit of the data in the absorbance vs. time plots (see insets in Fig. 5) yielded the initial photodecomposition rates. In both cases, the results for the experiments in Ar-purged solutions are similar to those under air atmosphere. Thus, no significant effect of the presence of oxygen in the photodegradation efficiency was observed.
The photodecomposition quantum yield, ΦR, was respectively determined for gadusol and gadusolate. For this purpose, acid solutions of gadusol were irradiated at 254 nm and neutral solutions at 303 nm. The quantum yield was calculated from the ratio of the initial photodecomposition rate and Ia, the absorbed radiation intensity. Ia was estimated by Io × (1 − 10−<A>) where <A> stands for the average absorbance of the solution at the irradiation wavelength. The incident radiation intensities Io for the 254 nm and the 303 nm excitation sources were determined by chemical actinometry and yielded respectively (3.70 ± 0.35) × 10−9 and (1.10 ± 0.03) ×10−8 einstein s−1. The results are shown in Table 1.
Laser flash photolysis studies
A significant bleaching was observed after a few cycles of laser pulses in the laser flash photolysis (LFP) experiments on acidic solutions of gadusol. On the contrary, the solutions were stable enough under neutral pH conditions to allow the study of the transient intermediates upon pulsed excitation of gadusolate. However, within the instrumental detection limit, no transient absorption was observed by direct LFP at 266 nm of aqueous gadusolate at pH 7.Thus, photosensitized formation of gadusolate triplet state was intended by energy transfer from various organic molecules that are known to produce high yields of triplet states in polar media: benzophenone, acridine, anthracene and rose bengal (RB).33
When benzophenone was photoexcited at 355 nm in methanol in the presence of gadusolate anion, the transient absorption spectrum 5 μs after the excitation clearly distinguished from the transient spectrum obtained without gadusol: an increasing band appears around 600 nm together with one band shifted towards ca. 340 nm and a minor one at 365 nm (see Fig. 6).34
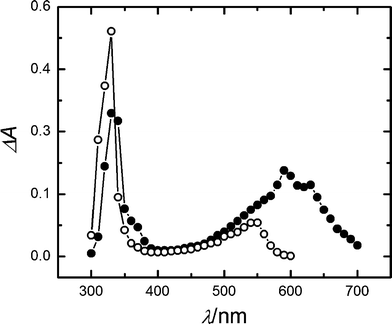 | ||
| Fig. 6 Transient absorption spectra from LFP experiences (λexc = 355 nm) in methanol, 5 μs after the laser pulse: 5 × 10−3 M benzophenone (open circles); 5 × 10−3 M benzophenone and 2 × 10−4 M gadusolate (full circles). | ||
Similarly, by LFP of acridine in the presence of gadusolate in methanol at 355 nm, a net increase of the absorption in the region of ca. 500 nm (data not shown) was observed some microseconds after the laser pulse. Simultaneously, the intensity and the lifetime of the signals around 440 nm, which are ascribed to the triplet–triplet absorption of acridine, shortened with the concentration of gadusolate.
However the transient absorption spectrum of anthracene in methanol (λexc = 355 nm) showed no change in the presence of gadusolate at concentrations as large as 2.3 × 10−3 M. Under these conditions, and considering the experimental lifetime of triplet anthracene in methanol (∼8 μs), the fraction of triplet states that can be intercepted by gadusolate is estimated to amount between 99 and 60%, by assuming a diffusion controlled quenching rate or up to a 102 factor slower.
Conversely, the transient absorption spectra in the microseconds of the LFP of RB at 532 nm (pH 7 buffer) were noticeably modified by the presence of gadusolate (Fig. 7).
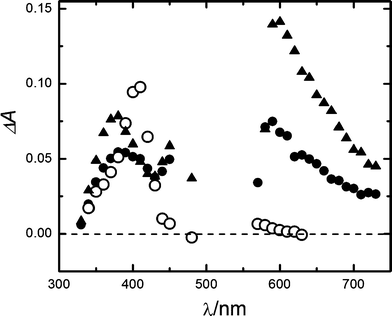 | ||
| Fig. 7 Transient absorption spectra from LFP experiences (λexc = 532 nm) in aqueous pH 7 buffer. 4 × 10−6 M RB, 4 μs (full triangles) and 50 μs (full circles) after the laser pulse; 4 × 10−6 M RB and 6 × 10−4 M gadusolate, 50 μs (open circles) after the laser pulse. | ||
Typical peak absorptions for RB triplet and RB radicals 4 and 50 μs after the laser pulse are distinguished at 380, 450 and 590 nm in the absence of gadusolate.35 However, the signal at 50 μs in the presence of 4 × 10−6 M gadusolate showed considerable differences: the bands at 450 and 590 nm extinguish whereas the absorbance around 410 nm increases. The time evolution of the absorbance changes (Fig. 8) shows a rapid raise at 410 nm followed by a slower increase during the subsequent microseconds that correlates with the decay at 590 nm reflecting the RB triplet depletion.
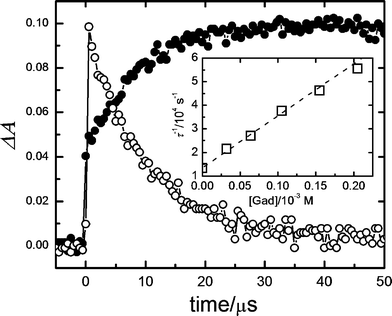 | ||
| Fig. 8 Absorbance profiles from the LFP (λexc = 532 nm) of 4 × 10−6 M RB in the presence of 6 × 10−4 M gadusol in aqueous solution (pH 7) at 410 nm (full circles) and 590 nm (open circles). Inset: Stern–Volmer plot for the quenching of RB triplet state by gadusolate in aqueous buffer (pH 7). τ stands for the RB triplet lifetime obtained from the absorbance decay at 600 nm (see text). | ||
The absorbance decays at 600 nm for different concentrations of gadusolate reflect a drop in τ, the RB triplet lifetime. The decays fit respectively to monoexponential functions of time from which the values of τ were determined. The data set was correlated in agreement with the Stern–Volmer expression (eqn (1)):
| τ−1 = τ0−1 + kq[Gad] | (1) |
Photoacoustic calorimetry studies
The non-radiative decay of excited-state gadusolate in water (pH 7) was assessed within the micro- and the early millisecond time ranges by photoacoustic calorimetry (PAC). The photoacoustic signals for gadusolate and potassium dichromate (the calorimetric reference) showed totally matched shapes (not shown here for simplicity) in aqueous media with equivalent thermoelastic properties.A linear dependence was verified for the plots of H (the signal amplitude) vs. laser fluence (Fig. 9). The slopes of the lines were taken as the energy-normalized signals Hn. The ratios of the Hn for gadusolate and the reference were respectively evaluated at two temperatures, 15 degrees apart from each other, in order to discriminate possible contributions to the photoacoustic signal from photoinduced structural volume changes.36 The fractions at both temperatures are coincident, within the experimental uncertainties, yielding approximately 1 (Table 2).
| Temperature | H Gadn | H Refn |
|
|---|---|---|---|
| 283 K | 0.063 ± 0.001 | 0.059 ± 0.001 | 1.066 ± 0.035 |
| 298 K | 0.124 ± 0.004 | 0.120 ± 0.003 | 1.030 ± 0.043 |
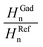
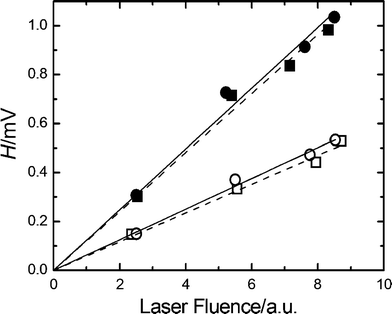 | ||
| Fig. 9 PAC signal amplitude, H, vs. the laser fluence for gadusolate (circles) and the calorimetric reference (squares) at 283 K (empty symbols) and 298 K (full symbols). Lines show the linear regression of the data. The slope of each line affords Hn, the value of the energy-normalized signal. | ||
Discussion
The solvatochromic shift in the UV absorption spectrum of aqueous gadusol on going from pH 2.5 to pH 7 (Fig. 1) has been explained in terms of the deprotonation of the enol form (gadusol) to give the resonance-stabilized enolate (gadusolate) that absorbs at lower energies.4,5 Consistently, our HPLC analysis reveals that only one peak with a distinctive retention time is observed at each pH, confirming that the protonation equilibrium is completely shifted towards one of the two mentioned species.6Moreover, the examination of the generalized solvatochromic shifts for the enol species enabled us to preliminarily assign the nature of the electronic transition in the photoexcitation process. The red shift in the absorption maximum on increasing polarity of the solvent (Fig. 2) (positive solvatochromism) accounts for a relative stabilization of its excited state with respect to the ground state that suggests the π–π* nature of the former.31,37
Furthermore, the results from the molecular calculations support this assignation of the excited state nature on the basis of the MOs involved in the most intense transition (Fig. 4), which are both compatible with the π-symmetry. Fig. 1 illustrates the dominance of this central transition in the simulated absorption band that completely overlaps the other two transitions at the sides. The HOMOs connected by the weaker absorptions show a strong non bonding character given by the electron density around the ketone oxygen (Fig. 4). Consistently, n→π* electronic transitions usually have low oscillator strengths due to symmetry selection rules, thus it seems reasonable to consider that nπ* excited states do not contribute significantly.
Besides, a closer inspection of Fig. 2 led us to propose that specific interactions may take place between gadusol and the solvent molecules that explain the departure from the linear trend, for example with 1,4-dioxane. Hydrogen bond (HB) donor/acceptor abilities of the molecules that contribute to the overall solvent polarity are taken into account by empirical approaches such as the solvatochromic comparison method by Kamlet and Taft et al.37,38 The multiparametric analysis of our results in terms of the polarizability and hydrogen donor/acceptor properties defined by these authors (Fig. 3) yielded a strong correlation for the complete set of solvents. The analytical description of the Kamlet–Taft solvatochromic relationship obtained on our data reflects a large contribution of the polarizability of the solvent, i.e., the π*-coefficient. Similar weights of the α- and β- terms in the equation indicate a comparable importance of the hydrogen bond donor and acceptor characters of gadusol–solvent interactions. The presence of hydroxylic and ketone functional groups in the chromophore of gadusol (Fig. 1) easily explains the ability for both kinds of HB interactions in the tested solvents.
The results summarized in Table 1 describe the high photostability of gadusol and gadusolate in aqueous solution, which appear not to be influenced by the presence of oxygen. Although the photodecomposition quantum yields are very low for both species, the values drop two orders of magnitude on going from the neutral molecular form to the anionic one. Thus, it is evident that the physical deactivation of the excited state of gadusolate is much faster than for gadusol, suggesting a less favoured competition of alternative reactive relaxation paths in the former case. Coincidentally, the most photostable species is that one prevailing at physiological proton concentrations. This has important consequences for the analysis of the photoprotection role of gadusol in the biological context, as discussed below.
Besides, the contribution of O2-mediated degradation steps to the reactivity of the excited states for both gadusol and its anion should be disregarded as the presence of oxygen has no effect on the photostability (Table 1).
No fluorescence emission was observed either, indicating that the main deactivation pathways for the excited states of gadusol and gadusolate are the non-radiative decays.
This is confirmed for gadusolate by the results of the PAC studies. The photoacoustic signal amplitude is a measure of the overall volume change induced in solution by the absorption of laser photons. Thus, two sources can be distinguished: (a) the thermal expansion of the solution due to the heat delivery in the non radiative relaxation processes, and (b) the molecular structural changes upon the electronic excitation of the species. The contributions can be separated by variation of the temperature since the thermal expansion depends strongly on this parameter.36 According to the data in Table 2, the ratio of signal amplitudes for gadusolate and the calorimetric reference remains invariable, within the experimental uncertainties, in the explored interval of temperatures. On this basis, it can be assumed that no net structural volume change occurs within the time window of the technique (ca. 1 μs),20 and that the signal is dominated by the thermal component. Besides, the data in the last column of Table 2 show that, within the experimental error, the ratio of energy-normalized signals remains around 1. This means that the relaxation behaviour of excited gadusolate is completely equivalent to that of the calorimetric reference, which promptly releases all the absorbed energy as heat to the surroundings within the first tens of ns.36
The result is also consistent with the negligible production of longer-lived intermediates (triplets) from singlet excited gadusolate demonstrated by the direct pulsed-laser photolysis at 266 nm.
It is known that cyclic α,β-unsaturated ketones produce relatively stabilized triplets when substituted at the β-carbon.39 From this viewpoint, it was expected to observe the formation of triplet state intermediates in the relaxation of gadusolate. Therefore, the lack of long-lived signals in the transient absorption experiments suggests the competition with extremely rapid non-reactive decays.
It is evident that the low yields of fluorescence, photodecomposition and intersystem crossing of gadusolate are a consequence of the very short lifetimes of the excited singlet states due to their rapid internal conversion. The allowed character of the π→π* transitions connecting the ground and first singlet states (see above) supports this view.
The photodecomposition quantum yield of mycosporine-glutaminol-glucoside, a natural analogous structure, has also been found to be remarkably low in aqueous solution.23 Similarly, previous results on the photostability and photophysical in vitro properties of the related imino-MAAs are in line with our findings for gadusol.18,20,21
From our results, it is confirmed that gadusol presents good UV-protective properties under physiological pH, which supports its sunscreening role, particularly relevant in the prebiotic environment. As biogenesis took place under very intense UV radiation, long before the formation of the stratospheric ozone layer, the incidence of an extreme selection pressure for UV protection in the molecular evolution has been suggested.40 Thus, it is probable that the development of photoprotection functionalities has involved gadusols and the related MAAs, on the basis of their advantageous photophysics.
The discussion about the molecular mechanisms of the photostability of life has frequently addressed the case of the nucleic acid bases which entail strongly comparable characteristics with gadusol such as: intense π→π* transitions, low fluorescence quantum yields and high stability towards photodegradation.41 The ultrafast non-radiative decay in nucleic acids has came into focus lately through femtosecond laser spectroscopy and modern theoretical photochemistry, which associates the efficiency of these relaxation processes to the crossings of the potential energy hypersurfaces of the electronic states known as conical intersections.41,425 Work in this direction should be carried out on gadusols and related compounds in order to provide more insight into the mechanism of the non-radiative relaxation.
On the other hand, the indirect photolysis experiments suggest that gadusolate may go through electron transfer processes in the presence of some triplet state sensitizers. The LFP experiment with benzophenone in methanol points to the formation of the sensitizer ion radical when gadusolate is present. The transient spectra in Fig. 6 include the typical bands of triplet benzophenone centred at 330 and 550 nm.43 The new bands in the region of 600 nm, 340 nm, and 365 nm, all appearing with the addition of gadusolate, are ascribed to the benzophenone radical anion.33,44
Analogously, the photosensitization with acridine evidenced the bleaching of its triplet–triplet absorption band at 440 nm in the presence of gadusolate and the development of a broad absorption around 500 nm, which coincides with that one ascribed to the neutral radical (“C” radical) from acridine.45 This neutral radical might has been produced from the electron transfer from gadusolate to acridine triplet, yielding the anion radical which is rapidly protonated in the alcoholic medium.46 Similar results have been reported for the reductive quenching of acridine-9-carboxylate in the presence of electron donors such as Trolox® or ascorbic acid.47
Thanks to the solubility of the sensitizer in water, the experiments with RB are particularly relevant for the examination of the electron transfer reactivity of gadusolate in aqueous solution. Semi-oxidized and semi-reduced forms of RB are known to be produced in the photolysis of RB as a consequence of the triplet–triplet annihilation and from the self-quenching reaction between triplet and ground states of RB.35 These ion radical species absorb between 380 and 470 nm, being the peak near 420 nm assigned to the anion radical of RB.48 Thus, the enhancement in this zone of the spectrum in the presence of gadusolate, concomitant with the decrease in the absorption due exclusively to RB triplet (ca. 590 nm) (Fig. 7), led us to propose the formation of semireduced RB by the electron transfer quenching of RB triplet by gadusolate. Consistent with this scheme, the time profile of the absorption at 590 nm in Fig. 8 decays almost monoexponentially, while the absorption at 410 nm increases in the microseconds range with the same time constant. The initial rapid raise in the signal at 410 nm is explained by the contribution to the absorption of RB triplet promptly produced upon laser excitation.
The time constant for the decay at 590 nm of RB triplet depends on gadusol concentration according to the Stern–Volmer kinetic model (see inset in Fig. 8), thus supporting a collisional mechanism of quenching. However, the derived value for the quenching constant kq ∼ 2 × 108 M−1s−1, indicates that the rate of the reductive quenching process is below the diffusion limit in water.
In agreement with the redox properties already reported for gadusols,2,5 we propose that gadusolate (Gad−) reacts with triplet sensitizer 3Sens*, as a reductive quencher according to (eqn (2)):
| Gad− + 3Sens* → Sens˙− + Gad˙ | (2) |
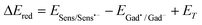 | (3) |
If we consider the reported data on the reduction potential for anthraceneESens/Sens˙− = −2.0 V and its triplet energy ET = 1.85 V,33 and compare them with the corresponding values for the rest of the sensitizers: acridine (ESens/Sens˙− = −1.56 V,50ET = 1.95 V); RB (ESens/Sens˙− = −0.78 V,51ET = 1.77 V) and benzophenone (ESens/Sens˙− = −1.72 V, ET = 3.00 V),33 it is clear that the case of anthracene yields the less favourable balance in terms of the spontaneity of the redox reaction.
This affinity of gadusolate towards reduction of photoexcited species is relevant in order to consider it as an additional mechanism supporting the antioxidant capacity of gadusols.6,27
Essential constituents of living cells such as riboflavin (Rb) and its derivatives may photosensitize harmful reactions, for example DNA damage. By considering the values for Rb in eqn (3): ESens/Sens˙− = −0.53 V and ET = 200 kJ mol−1,52 it can be predicted that triplet Rb is even easier to quench by gadusolate than RB triplet. Thus, the presence of gadusol may effectively contribute to the protection of these systems.
Experimental
Isolation and identification of gadusol
Gadusol was isolated from fish roe of Argentinian sandperch (Pseudopercis semifasciata). Details concerning the extraction and purification steps were described elsewhere.5,6 Briefly, the gonad’s content was homogenized in ethanol, processed by solvent extraction with chloroform and distilled water, and the aqueous phase treated by ion-exchange chromatography. The presence and concentration of gadusol in the solution samples were determined by the UV-absorption at λmax = 268 nm (pH 2–3).3,8 Reversible shift of the absorption maximum to 296 nm at pH ≥ 7 was taken into account as an additional criterion of fraction selection during the elutions.5,9 The purity of the samples was evaluated by reversed-phase HPLC on a Konik KNK-500-A system with UV-detection fixed at 268 nm. A 4.6 mm i.d. × 250 mm length, 5 μm C-18 ODS Aqua Phenomenex column and isocratic elution with 0.01% v/v acetic acid aqueous mobile phase at 1 mL min−1 flow-rate was used. Analysis by 1H-NMR spectroscopy was carried out for the confirmation of the chemical structure of gadusol.6Solutions were prepared by dilution with 0.5 M acetic acid (Cicarelli P.A. reagent) or with phosphate buffers of the gadusol samples eluted from the ion-exchange chromatography. Three times-distilled water (conductivity < 1.0 μS) was employed in all the cases. The phosphates for the buffers solutions were P.A. Cicarelli NaH2PO4 and Na2HPO4, and KH2PO4 (Sigma) and K2HPO4 (Fluka) for the LIOAS experiments.
Steady-state fluorescence and absorption measurements
Ground-state UV-visible absorption spectra of aqueous gadusol were recorded with a Shimadzu UV-2101 PC scanning spectrophotometer in 1 cm pathlength quartz cuvettes. Aqueous solutions of gadusol at pH 2.5 and 7 were prepared respectively with 0.5 M acetic acid and phosphate buffer.For the evaluation of the solvent effect on the absorption spectra, 10 μL aliquots of ca. 3 × 10−3 M gadusol in 0.5 M acetic acid aqueous solution were diluted in 2 mL of the solvent. The spectra were determined using neat solvent or the solvent mixture as the reference. The solvents used were: acetonitrile (Baker) and methanol (Mallinckrodt) both HPLC grade; acetic and propionic acids (Merck); ethanol, 2-propanol and 1,4-dioxane (Cicarelli), 1-propanol (Anedra), and 2-butanol (Raudo), all P.A. grade.
Corrected steady-state emission spectra at pH 2.5 and 7 were obtained on a Spex Fluoromax spectrofluorimeter at room temperature in 1 cm pathlength fluorescence quartz cuvettes. Absorbance of gadusol was ca. 0.30 at the excitation wavelength. Blank spectra were measured for each media in the absence of gadusol.
Photostability
Stationary photolysis of gadusol solutions were carried out by triplicate under air- and under high purity Ar-saturated atmospheres. Continuous illumination from a 4 W germicide mercury lamp (254 nm, Toshiba) was used for the photolysis at pH 2.5. For neutral or basic solutions, the UV-light source consisted of a 1000 W high-pressure Xe–Hg lamp attached to a grating monochromator in a Czerny–Turner mounting (Kratos-Schoeffel) with emission wavelength fixed at 303 ± 10 nm. Solutions with absorbance values between 0.5 and 0.7 at the irradiation wavelength were placed on 1 cm quartz cells under continuous stirring. Absorption spectra were recorded as a function of time. Blanks were carried out as control experiments in all the cases, probing that dark degradation was negligible. HPLC analyses on 20 μL aliquots of the irradiated mixture were also carried out at regular intervals of time during the photolysis.Reported molar absorption coefficients for gadusol (ε268 = 12400 and ε296 = 21800 M−1 cm−1)5 were considered to convert the absorbance to concentration units.
Phenylglyoxylic acid (Fluka), recrystallized from CCl4 (P.A. Cicarelli), was used as the chemical actinometer. The actinometer solutions were ca. 0.027 M in 3:1 acetonitrile–water solvent mixture. The photolysis rate of the phenylglyoxylic acid was determined by triplicate from the decrease in the absorbance at 380 nm (ε380 = 29 cm−1 M−1) as a function of time.53
Laser flash photolysis
Transient absorption measurements for the exploration of the triplet state and photosensitized reactions were carried out by LFP with the second, third or fourth harmonics of a Nd:YAG laser (Spectron Laser SL40, UK) as excitation sources.Further details concerning the LFP system are described elsewhere.54 Signals were obtained after averaging 10 laser pulses.
Direct photolysis experiments were performed by excitation at 266 nm on gadusol solutions (absorbance ca. 0.3 at 266 nm) previously deoxygenated by bubbling Ar (5.0) for 20 min.
Acridine (98% HPLC grade, Fluka), benzophenone (99%, Aldrich), anthracene (99.9% Aldrich, purified by sublimation) and RB (95%, Aldrich, without further purification) were used as sensitizers for the indirect photolysis experiments. The 355 nm output of the Nd:YAG laser was used as the excitation source in all these cases except for the 532 nm output used for RB. As before, all solutions were conveniently deoxygenated.
Aliquots of 4.3 × 10−3 M gadusol in buffer solution were added with a microsyringe to the sample cuvette for the quenching experiments.
Triplet lifetimes were obtained by fitting the absorbance decay signals to monoexponential functions.
Photoacoustic calorimetry (PAC)
The instrumental set-up has been already described.55 The laser beam from a Nd:YAG Spectron Laser SL400, operating at 266 nm, was circularly shaped with a 1.0 mm diameter pinhole. Photoacoustic signals were obtained as the average from 10 laser shots. Their amplitudes H were estimated from the vertical distance between the two first peaks in the detected pressure wave. H probed to be linear for both, sample and reference, with the incident laser fluence up to ca. 50 μJ.Potassium dichromate (recrystallized, P.A. Merck) was used as the calorimetric reference. Both, gadusol and potassium dichromate solutions were prepared in aqueous pH 7 buffer (0.1 M potassium mono- and di-hydrogenphosphate) in order to keep equivalent thermoelastic properties. Sample and reference absorbances at 266 nm were matched at around 0.290 ± 0.001. Working temperatures were 283.0 and 298.0 ± 0.2 K.
Molecular calculations
Density functional theory (DFT) and time-dependent density functional theory (TD-DFT) calculations were performed for Gadusol. Both methods were applied with the B3LYP hybrid functional approach by Gaussian 03W software.56,57 The geometry optimization of the ground state was carried out with the 6-31G* atomic basic set. Several other basis functions were tested by TD-DFT in order to reproduce the experimental absorption spectrum. The solvent effects were simulated with the polarizable continuum model (PCM) which assumes the solvent as a continuous dielectric medium and the solute molecule allocated in a cavity made of a series of spheres centred in each atom.58 The initial molecular geometry Z-matrix of the enol form of gadusol was generated by the version 8.0 Hyperchem software.Acknowledgements
This study was supported by CONICET (PIP5909), Universidad Nacional de Mar del Plata (15E-367) and Universidad Nacional de Río Cuarto (PPI 18C/256). E.M.A. thanks CONICET for a graduate studentship. M.S.C. and S.G.B. are research members of Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET, Argentina). The authors thank Dr M. A. Grela (UNMdP) for helpful suggestions regarding molecular calculations.References
- R. P. Sinha, S. P. Singh and D.-P. Häder, Database on mycosporines and mycosporine-like amino acids (MAAs) in fungi, cyanobacteria, macroalgae, phytoplankton and animals, J. Photochem. Photobiol., B, 2007, 89, 29–35 CrossRef CAS.
- J. M. Shick and W. C. Dunlap, Mycosporines-like amino acids and related gadusols: Biosynthesis, accumulation, and UV-protective functions in aquatic organisms, Annu. Rev. Physiol., 2002, 64, 223–262 CrossRef CAS.
- F. Chioccara, A. Della Gala, M. De Rosa, E. Novellino and G. Prota, Mycosporine aminoacids and related compounds from the eggs of fishes, Bull. Soc. Chim. Belg., 1980, 89, 1101–1106 CAS.
- P. T. Grant, P. A. Plack and R. H. Thomson, Gadusol, a metabolite from fish eggs, Tetrahedron Lett., 1980, 21, 4043–4044 CrossRef CAS.
- P. A. Plack, N. W. Fraser, P. T. Grant, C. Middleton, A. I. Mitchell and R. H. Thomson, Gadusol, an enolic derivative of cyclohexane-1,3-dione present in the roes of cod and other marine fish, Biochem. J., 1981, 199, 741–747 CAS.
- E. M. Arbeloa, M. J. Uez, S. G. Bertolotti and M. S. Churio, Antioxidant activity of Gadusol and occurrence in fish roes from Argentine Sea, Food Chem., 2010, 119, 586–591 CrossRef CAS.
- F. Chioccara, L. Zeuli and E. Novellino, Occurrence of mycosporine related compounds in sea urchin eggs, Comp. Biochem. Physiol., Part B: Biochem. Mol. Biol., 1986, 85, 459–461 CrossRef.
- P. T. Grant, C. Middleton, P. A. Plack and R. H. Thomson, The isolation of four aminocyclohexenimines (mycosporines) and a structurally related derivative of cyclohexane-1,3-dione (Gadusol) from the brine shrimp Artemia, Comp. Biochem. Physiol., Part B: Biochem. Mol. Biol., 1985, 80, 755–759 CrossRef.
- W. M. Bandaranayake, D. J. Bourne and R. G. Sim, Chemical composition during maturing and spawning of the sponge Dysidea herbacea (Porifera: Demospongiae), Comp. Biochem. Physiol., Part B: Biochem. Mol. Biol., 1997, 118, 851–859 CrossRef.
- W. M. Bandaranayake, Mycosporines. Are they nature's sunscreens?, Nat. Prod. Rep., 1998, 15, 159–172 RSC; R. P. Rastogi, R. R. P. Sinha, S. P. Singh and D-P. Haëder, Photoprotective compounds from marine organisms, J. Ind. Microbiol. Biotechnol., 2010, 37, 537–558 CrossRef CAS.
- D. L. Fabacher, E. E. Little and G. K. Ostrander, Tolerance of an albino fish to ultraviolet-B radiation, Environ. Sci. Pollut. Res., 1999, 6, 69–71 CrossRef CAS.
- A. I. Callone, M. Carignan, N. G. Montoya and J. I. Carreto, Biotransformation of mycosporine like amino acids (MAAs) in the toxic dinoflagellate Alexandrium tamarense, J. Photochem. Photobiol., B, 2006, 84, 204–212 CrossRef CAS.
- J. Favre-Bonvin, J. Bernillon, N. Salin and N. Arpin, Biosynthesis of mycosporines: mycosporine glutaminol in Trichothecium roseum, Phytochemistry, 1987, 26, 2509–2514 CrossRef CAS.
- J. M. Shick, S. Romaine-Lioud, C. Ferrier-Pagès and J.-P. Gattuso, Ultraviolet-B radiation stimulates shikimate pathway-dependent accumulation of mycosporine-like amino acids in the coral Stylophora pistillata despite decreases in its population of symbiotic dinoflagellates, Limnol. Oceanogr., 1999, 44, 1667–1682 CAS.
- W. C. Dunlap and J. M. Shick, Ultraviolet radiation-absorbing mycosporine-like amino acids in coral reef organisms: a biochemical and environmental perspective (Review), J. Phycol., 1998, 34, 418–430 CrossRef.
- J. M. Shick, W. C. Dunlap and G. R. Buettner, Ultraviolet (UV) protection in marine organisms II. Biosynthesis, accumulation, and sunscreening function of mycosporine-like amino acids, in Free Radicals in Chemistry, Biology and Medicine, ed. T. Yoshikawa, S. Y. Toyokuny Yamamoto and Y. Naito, OICA International, London, 2000, pp. 215–228 Search PubMed.
- F. García-Pichel, C. E. Wingard and R. W. Castenholz, Evidence regarding the UV sunscreen role of a mycosporine-like compound in the cyanobacterium Gloeocapsa sp., Appl. Environ. Microbiol., 1993, 59, 170–176 CAS.
- F. R. Conde, M. S. Churio and C. M. Previtali, The photoprotector mechanism of mycosporine-like amino acids. Excited-state properties and photostability of porphyra-334 in aqueous solution, J. Photochem. Photobiol., B, 2000, 56, 139–144 CrossRef CAS.
- H.-H. Perkampus, in UV-VIS Atlas of organic compounds, ed. VCH, Weinheim, 2nd edn, 1992, Part 1 Search PubMed.
- F. R. Conde, M. S. Churio and C. M. Previtali, The deactivation pathways of the excited-states of the mycosporine-like amino acids shinorine and porphyra-334 in aqueous solution, Photochem. Photobiol. Sci., 2004, 3, 960–967 RSC.
- F. R. Conde, M. S. Churio and C. M. Previtali, Experimental study of the excited-state properties and photostability of the MAA palythine in aqueous solution, Photochem. Photobiol. Sci., 2007, 6, 669–674 RSC.
- K. Whitehead and J. I. Hedges, Photodegradation and photosensitization of mycosporine-like amino acids, J. Photochem. Photobiol., B, 2005, 80, 115–121 CrossRef CAS.
- M. Moliné, E. M. Arbeloa, M. R. Flores, D. Libkind, M. E. Farías, S. G. Bertolotti, M. S. Churio and M. R. van Broock, UVB Photoprotective Role of Mycosporines in Yeast: Photostability and Antioxidant Activity of Mycosporine-Glutaminol-Glucoside, Radiat. Res., 2011 DOI:10.1667/RR2245.1.
- W. C. Dunlap and Y. Yamamoto, Small-molecule antioxidants in marine organisms: antioxidant activity of microsporine-glycine, Comp. Biochem. Physiol., Part B: Biochem. Mol. Biol., 1995, 112, 105–114 CrossRef.
- F. de la Coba, J. Aguilera, F. L. Figueroa, M. V. de Gálvez and E. Herrera, Antioxidant activity of mycosporine-like amino acids isolated from three red macroalgae and one marine lichen, J. Appl. Phycol., 2009, 21, 161–169 Search PubMed.
- W. C. Dunlap, J. M. Shick and Y. Yamamoto, Ultraviolet (UV) protection in marine organisms l. Sunscreens, oxidative stress and antioxidants, in Free radicals in chemistry, biology and medicine, ed. S. Yoshikawa, S. Toyokuni, Y. Yamamoto and Y. Naito, OICA International, London, 2000, pp. 201–214 Search PubMed.
- H.-J. Suh, H.-W. Lee and J. Jung, Singlet oxygen quenching by deoxygadusol and related mycosporine-like amino acids from phytoplankton Prorocentrum micans, J. Photosci., 2004, 11, 77–81 Search PubMed.
- J. Rozema, L. O. Björn, J. F. Bornman, A. Gaberščik, D.-P. Häder, T. Trošt, M. Germ, M. Klisch, A. Gröniger, R. P. Sinha, M. Lebert, Y.-Y. He, R. Buffoni-Hall, N. V. J. de Bakker, J. van de Staaij and B. B. Meijkamp, The role of UV-B radiation in aquatic and terrestrial ecosystems—an experimental and functional analysis of the evolution of UV-absorbing compounds, J. Photochem. Photobiol., B, 2002, 66, 2–12 CrossRef CAS.
- F. Garcia-Pichel, Solar ultraviolet and evolutionary history of cyanobacteria, Origins Life Evol. Biosphere, 1998, 28, 321–347 CrossRef CAS.
- C. S. Cockell and J. Knowland, Ultraviolet radiation screening compounds, Biol. Rev., 1999, 74, 311–345 CrossRef CAS.
- C. Reichardt, Solvatochromic dyes as solvent polarity indicators, Chem. Rev., 1994, 94, 2319–2358 CrossRef CAS.
- J. L. Jones and S. C. Rutan, Solvatochromic studies of stationary phases on thin-layer chromatographic plates, Anal. Chem., 1991, 63, 1318–1322 CrossRef CAS.
- S. L. Murov, I. Carmichael and G. L. Hug, in Handbook of Photochemistry, ed. Marcel Dekker Inc., New York, 2nd edn, 1993 Search PubMed.
- T. Shida, Electronic Absorption Spectra of Radical ions, Elsevier Science Pub., Amsterdam and New York, 1988 Search PubMed.
- V. Wintgens, J. C. Scaiano, S. M. Linden and D. C. Neckers, Transient phenomena in the laser flash photolysis of rose Bengal C-2′ ethyl ester C-6 sodium salt, J. Org. Chem., 1989, 54, 5242–5246 CrossRef CAS.
- S. E. Braslavsky and G. E. Heibel, Time-resolved photothermal and photoacoustic methods applied to photoinduced processes in solution, Chem. Rev., 1992, 92, 1381–1410 CrossRef CAS; T. Gensch and C. Viappiani, Time-resolved photothermal methods: accessing time-resolved thermodynamics of photoinduced processes in chemistry and biology, Photochem. Photobiol. Sci., 2003, 2, 699–721 RSC.
- C. Reichardt, in Solvents and solvent effect in organic chemistry, ed. VCH, Weinheim, 2nd edn, 1990 Search PubMed.
- M. J. Kamlet, J. L. Abboud and R. W. Taft, The solvatochromic comparison method. 6. The π* scale of solvent polarities, J. Am. Chem. Soc., 1977, 99, 6027–6038 CrossRef CAS.
- D. I. Schuster, D. A. Dunn, G. E. Heibel, P. B. Brown, J. M. Rao, J. Woning and R. Bonneau, Enone photochemistry. Dynamic properties of triplet excited states of cyclic conjugated enones as revealed by transient absorption spectroscopy, J. Am. Chem. Soc., 1991, 113, 6245–6255 CrossRef CAS; J. M. Kelly, T. B. H. McMurry and D. N. Work, Laser flash photolysis of 3-phenylcyclopent-2-enone: Absorption spectrum and reactivity of its triplet excited state, J. Chem. Soc., Chem. Commun., 1987, 280–281 RSC; J. M. Kelly, T. B. H. McMurry and D. N. Work, Photochemistry of substituted cyclic enones. Part 7. Flash photolysis of 3-phenylcyclopent-2-enones and 3-phenylcyclohex-2-enone, J. Chem. Soc., Perkin Trans. 2, 1990, 981–983 RSC.
- C. S. Cockell and G. Horneck, The history of the UV radiation climate of the Earth—Theoretical and space-based observations, Photochem. Photobiol., 2001, 73, 447–451 CrossRef CAS; A. Y. Mulkidjanian, D. A. Cherepanov and M. Y. Galperin, Survival of the fittest before the beginning of life: selection of the first oligonucleotide-like polymers by UV light, BMC Evol. Biol., 2003, 3–12 Search PubMed.
- C. E. Crespo-Hernández, B. Cohen, P. M. Hare and B. Kohler, Ultrafast excited-state dynamics in nucleic acids, Chem. Rev., 2004, 104, 1977–2019 CrossRef CAS.
- S. Perun, A. L. Sobolewski and W. Domcke, Conical intersections in thymine, J. Phys. Chem. A, 2006, 110, 13238–13244 CrossRef CAS; L. Serrano-Andrés and M. Merchán, Are the five natural DNA/RNA base monomers a good choice from natural selection? A photochemical perspective, J. Photochem. Photobiol., C, 2009, 10, 21–32 CrossRef CAS.
- R. V. Bensasson and J.-C. Gramain, Benzophenone triplet properties in acetonitrile and water. Reduction by lactams, J. Chem. Soc., Faraday Trans. 1, 1980, 76, 1801–1810 RSC.
- A. Beckett and G. Porter, Primary photochemical processes in aromatic molecules, Trans. Faraday Soc., 1963, 59, 2038–2049 RSC.
- A. Kellmann and F. Tfibel, Radicals produced from the laser-induced photoionization of acridine in solution, J. Photochem., 1982, 18, 81–88 CrossRef CAS.
- P. Neta, Electron transfer reactions involving acridine and related compounds, J. Phys. Chem., 1979, 83, 3096–3101 CrossRef CAS.
- W. E. Ford and M. A. J. Rodgers, Acridine-9-carboxylate as an efficient triplet-state mediator of photosensitized electron-transfer reactions via a reductive quenching pathway, J. Phys. Chem., 1991, 95, 5827–5831 CrossRef CAS.
- C. Lambert, T. Sarna and T. G. Truscott, Rose Bengal radicals and their reactivity, J. Chem. Soc., Faraday Trans., 1990, 86, 3879–3882 RSC.
- B. H. J. Bielski, A. O. Allen and H. A. Schwarz, Mechanism of disproportionation of ascorbate radicals, J. Am. Chem. Soc., 1981, 103, 3516–3518 CrossRef; B. Halliwell and J. M. C. Gutteridge, Free Radicals in Biology and Medicine, 1999, Oxford Univ. Press, Oxford, 3rd edn, ch. 3 Search PubMed.
- J. S. Jaworski and M. Cembor, Kinetics of protonation of the acridine radical anion in DMF by water and alcohols, Tetrahedron Lett., 2000, 41, 7267–7270 CrossRef CAS.
- C. R. Lambert and I. E. Kochevar, Electron transfer quenching of the Rose Bengal triplet state, Photochem. Photobiol., 1997, 66, 15–25 CrossRef CAS.
- D. Meisel and P. Neta, One-electron reduction potential of riboflavin studied by pulse radiolysis, J. Phys. Chem., 1975, 79, 2459–2461 CrossRef CAS; P. F. Heelis, The photophysical and photochemical properties of flavins (isoalloxazin), Chem. Soc. Rev., 1982, 11, 15–39 RSC.
- A. Defoin, R. Defoin-Straatmann, K. Hildebrand, E. Bittersmann, D. Kreft and H. J. Kuhn, A new liquid phase actinometer: quantum yield and photo-CINDP study of phenylglyoxylic acid in aqueous solution, J. Photochem., 1986, 33, 237–255 CrossRef CAS.
- S. G. Bertolotti and C. M. Previtali, The excited states quenching of safranine T by p-benzoquinones in polar solvents, J. Photochem. Photobiol., A, 1997, 103, 115–119 CrossRef CAS.
- M. A. Brusa, M. S. Churio, M. A. Grela, S. G. Bertolotti and C. M. Previtali, Reaction volume and reaction enthalpy upon aqueous peroxodisulfate dissociation: S2O82−→2SO4−, Phys. Chem. Chem. Phys., 2000, 2, 2383–2387 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, GAUSSIAN 03 (Revision C.01), Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
- A. D. Becke, Density–functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS; A. D. Becke, Density-functional exchange-energy approximation with correct asymptotic behaviour, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS; C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B: Condens. Matter, 1988, 37, 785–789 CrossRef CAS.
- S. Miertuš, E. Scrocco and J. Tomasi, Electrostatic interaction of a solute with a continuum. A direct utilization of ab initio molecular potentials for the prevision of solvent effects, Chem. Phys., 1981, 55, 117–129 CrossRef CAS.
Footnote |
| † In the following, we will use the term gadusolate to denote the deprotonated (enolate) gadusol species that prevails under neutral and alkaline conditions in aqueous solution. |
| This journal is © The Royal Society of Chemistry and Owner Societies 2011 |