Stereoselective interaction of ketoprofen enantiomers with β-cyclodextrin: ground state binding and photochemistry
Giancarlo
Marconi
a,
Elisabetta
Mezzina
b,
Ilse
Manet
a,
Francesco
Manoli
a,
Barbara
Zambelli
c and
Sandra
Monti
*a
aIstituto per la Sintesi Organica e la Fotoreattività, Consiglio Nazionale delle Ricerche, via Piero Gobetti 101, I-40129, Bologna, Italy
bDipartimento di Chimica Organica «A. Mangini», Università di Bologna, Viale Risorgimento 4, I-40126, Bologna, Italy
cDipartimento di Scienze e Tecnologie Agro-ambientali, Università di Bologna, Via Fanin 40, I-40127 Bologna, Italy and CERM (Center for Magnetic Resonance), University of Florence, Via Luigi Sacconi 6, I-50019, Sesto Fiorentino, Italy
First published on 26th October 2010
Abstract
The chiral recognition ability of β-cyclodextrin (β-CyD) vs.S- and R-ketoprofen (KP) enantiomers has been studied by circular dichroism (CD), isothermal titration calorimetry (ITC) and NMR. The association constants of the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complexes obtained from CD and ITC titration experiments resulted to be the same for both enantiomers within the experimental uncertainty. Well differentiated CD spectra were determined for the diastereomeric complexes. Their structure was assessed by molecular mechanics and molecular dynamics calculations combined with quantum mechanical calculation of the induced rotational strengths in the low energy KP:β-CyD associates, upon comparison of the calculated quantities with the corresponding experimental CD. The inclusion geometry is similar for both enantiomers with the aromatic carbonyl inserted in the CyD cavity, the monosubstituted ring close to the primary CyD rim and the carboxylate group exposed to the solvent close to the secondary rim. NMR spectra fully confirmed the geometry of the diastereomeric complexes. Tiny structural differences were sensibly probed by CD and confirmed by 2D ROESY spectra. Photoproduct studies with UV absorption and MS detection as well as nanosecond laser flash photolysis evidenced lack of chiral discrimination in the photodecarboxylation of KP within the cavity and formation of a photoaddition product to β-CyD by secondary photochemistry of 3-ethylbenzophenone.
:1 complexes obtained from CD and ITC titration experiments resulted to be the same for both enantiomers within the experimental uncertainty. Well differentiated CD spectra were determined for the diastereomeric complexes. Their structure was assessed by molecular mechanics and molecular dynamics calculations combined with quantum mechanical calculation of the induced rotational strengths in the low energy KP:β-CyD associates, upon comparison of the calculated quantities with the corresponding experimental CD. The inclusion geometry is similar for both enantiomers with the aromatic carbonyl inserted in the CyD cavity, the monosubstituted ring close to the primary CyD rim and the carboxylate group exposed to the solvent close to the secondary rim. NMR spectra fully confirmed the geometry of the diastereomeric complexes. Tiny structural differences were sensibly probed by CD and confirmed by 2D ROESY spectra. Photoproduct studies with UV absorption and MS detection as well as nanosecond laser flash photolysis evidenced lack of chiral discrimination in the photodecarboxylation of KP within the cavity and formation of a photoaddition product to β-CyD by secondary photochemistry of 3-ethylbenzophenone.
Introduction
Ketoprofen (KP) is a non-steroidal anti-inflammatory drug with a 2-arylpropionic acid core (Scheme 1). As with many molecules of this family, KP provokes photosensitizing side reactions due to the presence of the diarylketone moiety.1 In clinical practice the drug is administered as a racemic mixture even though it possesses enantiodifferentiated anti-inflammatory and analgesic activities.2 The photoreactivity mechanisms have been extensively investigated for racemic KP in various media.3–7 The role of the drug stereochemistry has been studied in serum albumin (SA) matrices. Stereodifferentiation does not manifest in the association constants, but is clearly evident in the circular dichroism (CD) spectra and in the photodegradation quantum yields of the drug protein complexes.8–11 Information on the nature of the drug:protein diastereomeric interactions has been obtained for bovine serum albumin from the combined application of isothermal titration calorimetry (ITC), CD spectroscopy and molecular mechanics (MM) and molecular dynamics (MD) simulations.12 Calculation of the induced rotational strengths of electronic transitions in low energy drug:protein associates by quantum mechanical methods and comparison of the calculated quantities with experimental induced CD (ICD) signals has proven to be a reliable method to assign the most likely structure to the diastereomeric complexes.13–15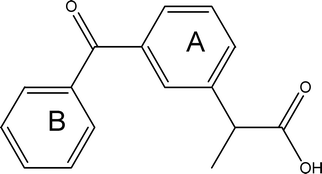 | ||
| Scheme 1 2-(3-Benzoylphenyl)propionic acid (ketoprofen, KP). | ||
Chiral discrimination by cyclodextrin (CyD) derivatives has been extensively investigated and exploited in chromatography for resolution of racemates16 and in the pharmaceutical field.17 The recognition power of CyDs is related to their inherent optical dissymmetry and their ability to host racemic guests in their hydrophobic cavities, forming complexes of diastereodifferentiated stability. Compared to proteins or to multiply substituted CyDs, natural CyDs are expected to be quite modest chiral selectors, essentially because the macrocycle is characterized by a high degree of geometrical symmetry and/or there is little interaction between the host asymmetric centers and the guest.18 In this frame the interaction of enantiomeric KP with CyDs has received some attention.19–24 The main goals of these studies were enantiomeric purity determinations and development of chiral separation procedures. To the best of our knowledge little has been done to understand the recognition process from the structural point of view and verify the possible existence of enantioselectivity in the photochemical reactions of KP in the CyD complexes.
The chiral center of KP is peripheral with respect to the benzoyl-phenyl moiety entering the β-CyD cavity,5,25 thus the structural differences between the diastereomeric KP:β-CyD complexes was expected to be small. We studied the association process of the individual KP enantiomers to β-CyD with CD, NMR and isothermal titration calorimetry (ITC). We determined equilibrium constants, thermodynamic parameters and absolute CD spectra of the complexes. The structures of the complexes were derived from NMR and the CD-based spectroscopic/computational approach mentioned above and already applied successfully to determine the inclusion geometry of several chiral CyD guests.26–28 We checked for the existence of diastereoselectivity in the photochemical behaviour of the KP complexes using laser flash photolysis and mass spectrometry. We had the opportunity to compare the various techniques for their ability to evidence tiny stereochemical effects.
Experimental
Materials
S-(+)-2-(3-Benzoylphenyl)propionic acid (ketoprofen, KP), was purchased from Sigma-Aldrich, Milan, Italy. R-(−)-2-(3-Benzoylphenyl)propionic acid preparation was already described.9 0.1 or 0.01 M phosphate buffer at pH 7.4 was used as solvent. Water was purified by passage through a Millipore MilliQ system (Millipore SpA, Milan, Italy). KP is present as the carboxylate anion at neutral pH (pKa = 4.7).1 The KP samples were carefully protected from ambient light during the manipulations in order to avoid photodegradation, occurring in aqueous solutions at neutral pH with very high quantum yields.29β-Cyclodextrin from Serva was used as received.Spectroscopic study of binding
Circular dichroism spectra were taken on a Jasco J-715 spectropolarimeter and absorption spectra on a Perkin Elmer λ9 spectrophotometer. Titration experiments were carried out at constant KP concentration (6 × 10−5 M) and twelve β-CyD concentrations. Complex stoichiometry and association constants were determined using a multiwavelength data analysis program (SPECFIT/32 by Spectrum Software Associates, TgK Scientific). The procedure, already described, was completely numerical, i.e. did not involve parametric equations.8,9,11–15 The analysis also afforded the individual spectra of the complexes.Complex structure and CD calculations
Molecular mechanics calculations were carried out for the complexation of the carboxylate anion of KP with β-CyD using the Consistent Valence Force Field (CVFF) included in the Discover module of the software InsightII,30 adopting the conjugate gradient method for convergence with a threshold of 4 × 10−3 kJ mol−1 Å−1 on the energy gradient. Four starting geometries were considered for docking of each KP enantiomer into β-CyD: entrance through primary or secondary rim, entrance of the aryl-ketone moiety with mono-substituted or di-substituted ring. The initially obtained MM minima structures were allowed to relax into the energetically more favourable conformations in a series of simulated annealing cycles until no significant conformational changes were observed in the following cycles. The molecular dynamics simulations were performed at 300 K and lasted for 1 ns, with time steps of 10 fs. A continuum solvation model was included in the calculations. The error on the binding energies was estimated to be ±0.7 kcal mol−1.31The circular dichroism (CD) of the complexes with lowest energy was calculated following a procedure based on the Tinoco's model for interacting groups of a polymer.32 According to this formulation, the rotational strength of an electronic transition 0→a is given by:
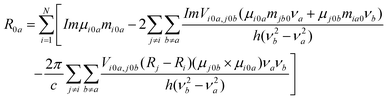 | (1) |
Microcalorimetric measurements
Isothermal titration calorimetry (ITC) was performed on a Microcal VP-ITC microcalorimeter. Data acquisition, instrument control and data analysis were carried out using the dedicated Origin software package (Microcal). All the experiments were performed at 25 °C with initial KP concentration of 1.2 × 10−3 M in the sample cell (1.400 mL). The reference cell was filled with deionized water. 30 aliquots of 10 μL of the CyD solution 1.2 × 10−2 M were injected from the syringe (300 μL) into the stirred sample cell (stirring speed 290 rpm). The syringe was recharged after 30 additions and further 30 aliquots were added. The KP and the CyD were diluted in the same buffer solution (0.1 M phosphate buffer at pH 7.4) in order to minimize non-specific heat of buffer mix. Use of higher concentrations of KP and CyD was not possible due to dimerization/solubility problems. A 300 s time interval between injections was applied in order to allow the system to reach thermal equilibrium after each addition. Experiments for CyD dilution heats were performed by titrating the CyD solution in the buffer alone under the same experimental conditions. Correction of data was performed for the heat of CyD dilution. Experimental data were fitted using a non-linear least-squares minimization algorithm to a theoretical titration curve. The enthalpy change of the binding reaction (ΔH), the affinity constant (K) and the number of sites (n) were the floating parameters of the fit. The best fit was found for a model involving a single set of sites (fitting parameters n, K, ΔH). The reaction entropy was calculated using the equations ΔG = −RTlnK (R = 1.9872 cal mol−1 K−1, T = 298 K) and ΔG = ΔH − TΔS.
Steady state photolysis
Solutions of R-KP and S-KP (1.0 × 10−4 M) and β-CyD (1.0 × 10−2 M) were prepared in triply distilled water of pH 7.4 adjusted with NaOH. The solutions were degassed with a gentle argon flow for 20 min and irradiated in 3 mL quartz cuvettes of 1 cm pathlength with light of a medium pressure mercury arc lamp in the presence of a cut off filter at 305 nm. In front of the sample a water filter was positioned in order to avoid heating of the solution. The sample was continuously stirred during the irradiation. The absorbance at 259 nm was checked at different irradiation times in order to monitor the early stages of the photobleaching of the π,π* band (for Δtirr < 2 min ΔA259 was < 5%). The experiments were performed in triplicate. Photoproducts were analyzed with ESI-mass spectrometry (see below) for solutions of KP (1.0 × 10−3 M) and β-CyD (1.0 × 10−2 M) (KP > 85% complexed) irradiated up to ∼3% photobleaching of the 259 nm band. They were also checked after ∼20% photobleaching. 10 times dilution of the irradiated solutions was performed with a mixture of water–MeOH (8/2), immediately before injection. Control experiments were performed for the β-CyD sample and the non irradiated mixture.Mass spectrometry
The instrument was a Bruker ion trap mass spectrometer with ESI source. Injection rate was 10 μl min−1. We optimized the mass ion trap for two different m/z values, 250 m/z and 1300 m/z in order to observe the drug alone as well as its β-CyD complexes.Nanosecond laser flash photolysis
Transient absorption spectra were taken with excitation at 355 nm from a Nd-YAG laser (JK-Lasers, 20 ns FWHM), suitably shaped to pass through a rectangular, 3 mm high and 10 mm wide window, providing a fairly uniform energy density incident onto the sample cell (3.8 mJ corresponding to 12.6 mJ cm−2). A front portion of 2 mm of the excited solution was probed at right angle, the useful optical path being 10 mm. A355 was ∼0.1 over 1 cm. Ar-saturated solutions were used. Temperature was 295 K. The sample was renewed after a few laser shots.NMR measurements
All 1D and 2D NMR spectra were recorded at 298 K on Varian Inova spectrometer operating at 600 MHz. NMR experiments were performed in D2O solutions using residual HOD as an internal standard (4.76 ppm). Chemical shifts are reported in parts per million (δ scale). ROESY data were carried out using a 90° pulse width of 6.5 μs and a spectral width of 6000 Hz in each dimension. The data were recorded in the phase sensitive mode using a CW spinlock field of 2 kHz, without spinning the sample. Acquisitions were recorded at mixing times 300 ms. Other instrumental settings are: 64 increments of 2 K data points, 8 scans per t1, 1.5 s delay time for each scan.Results
Circular dichroism
The intrinsic CD of S-KP and R-KP in phosphate buffer at pH 7.4 is extremely weak both in the 340–350 nm and 260 nm regions, corresponding to the n,π* S1←S0 and π,π* S2←S0 transitions, respectively. A signal positive for R- and negative for S-KP is present at ca. 220 nm. The CD signal of both the n,π* and the π,π* band becomes one order of magnitude more intense in the presence of β-CyD, indicating that the optical asymmetry of the environment and the conformational constraints induce substantial rotational strength in the drug electronic transitions. Fig. 1a and 1b show the effect of increasing β-CyD concentration on the ellipticity of a 6 × 10−5 M S-KP and R-KP solution, respectively. The CD variations were perfectly accounted for by the formation of 1:1 stoichiometry complexes with binding constants log(KS/M−1) = 3.04 ± 0.07 for S-KP and log(KR/M−1) = 2.88 ± 0.04 for R-KP. The absolute CD spectra are clearly diastereodifferentiated (Fig. 1d). The CD features of the diastereomeric complexes in the π,π* band agree with those reported in ref. 23, while those of the n,π* band were not reported before to be of opposite sign. The absorption spectra obtained from the analysis of the absorption titration appear quite similar (Fig. 1c).
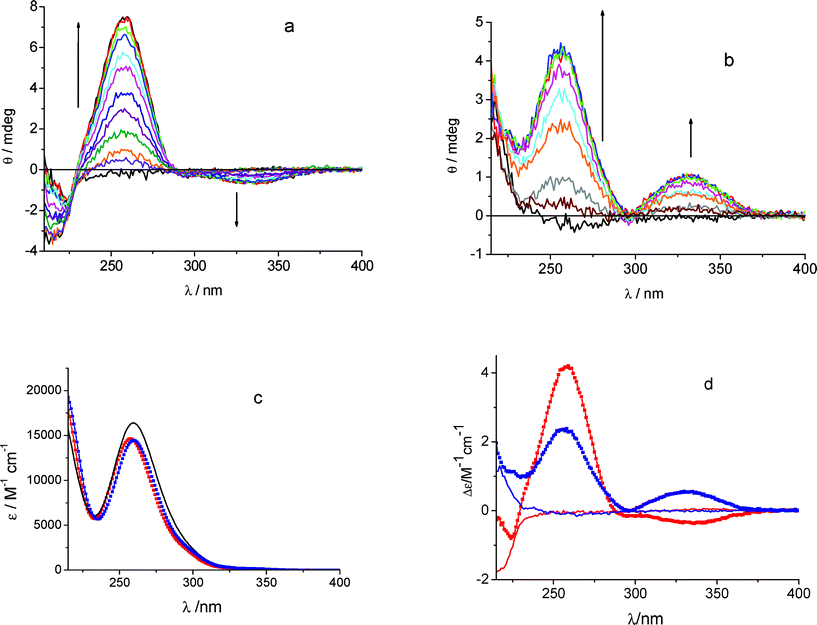 | ||
| Fig. 1 Ellipticity changes (Δθ) obtained titrating KP 6 × 10−5 M with β-CyD in the range 2 × 10−5–1.2 × 10−4 mol dm−3 in phosphate buffer 0.01 mol dm−3 pH 7.4, 25 °C, cell 1 cm (the signal of β-CyD alone was subtracted): (a) S-KP, (b) R-KP. (c) Comparison of the absorption spectra of the complexes of R-KP (blue) and S-KP (red) with the absorption spectrum of the pure KP enantiomers (black line). (d) Calculated CD spectra of the diastereoselective complexes (squares) of R-KP (blue) and S-KP (red) obtained from analysis of data in (a) and (b), respectively, together with the CD spectra of the pure enantiomers (lines). | ||
Isothermal titration calorimetry
ITC measurements were carried out at 25 °C by adding β-CyD to enantiomeric KP 1.2 × 10−3 M solutions. The concentration of KP was chosen to be as high as possible taking into account of solubility limits, but lower than that of NMR to avoid significant KP dimerization (see below, NMR measurements). The occurrence of a binding event was revealed by the presence of exothermic peaks following each addition (Fig. 2, top). Fits of the integrated heat data were carried out with a single site binding model. The best fit (Fig. 2, bottom) revealed that one CyD molecule bound to one molecule of enantiomeric KP (nS = 1.22, nR = 1.06) with similar affinity constants for both enantiomers (KS = 791 ± 19 M−1 and KR = 568 ± 8 M−1). The enthalpic factors were favorable, ΔHS = −3014 ± 59 cal mol−1 and ΔHR = −3728± 64 cal mol−1, while calculated entropic values were small and positive, ΔSS = 3.15 ± 0.25 cal mol−1 K−1 and ΔSR = 0.10 ± 0.24 cal mol−1 K−1. The large heat effect produced by CyD-KP interaction leads to very good signal-to-noise ratio and excellent fit of the thermodynamic data with small mathematical errors in the best fitting parameters. However, because of the relatively low KP concentration coupled to the relatively low binding affinity we were not in the optimal range of the critical parameter c = K × Mtot × n, where K is the calculated binding constant, Mtot is the KP concentration in the cell and n is the stoichiometry parameter. The c parameter determines the shape of the binding isotherm and should be 1 < c < 1000. When c < 1, as in our case, the drop near the equivalence point of the curve, which is correlated to the affinity constant, becomes broadened and the intercept at the Y axis, which is indicative of the ΔH value, tends to become lower than the true ΔH. Thus at the c values used in our experiments ITC cannot be so accurate to distinguish a K difference less than 2-fold and ΔH values so similar, thereby being unable to prove chiral discrimination.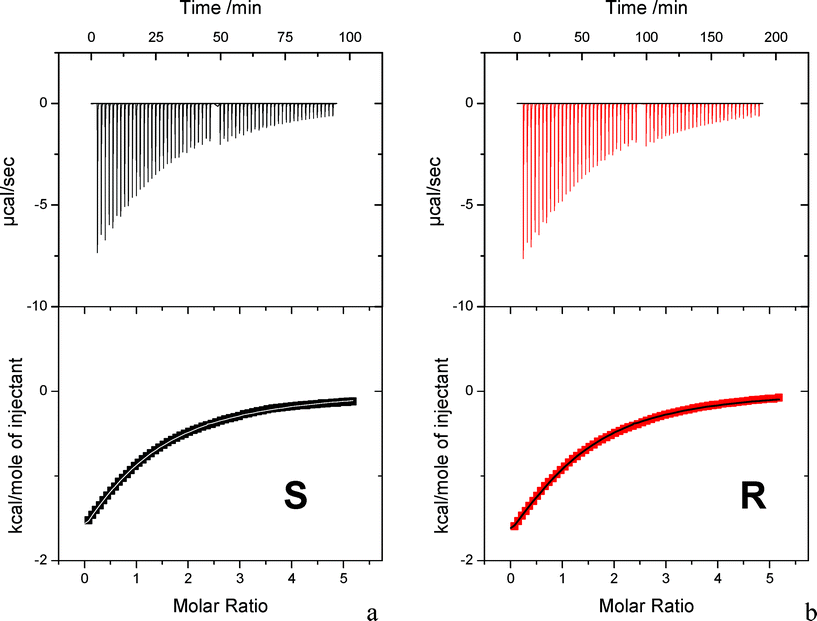 | ||
| Fig. 2 Calorimetric data for the titration of S-KP and R-KP with β-CyD at 25 °C. Heat output associated with each injection of drug as a function of time. In the higher molar ratio region the syringe was recharged and the experiment prosecuted; additional data were suitably normalized for insertion in the bottom plot. (Bottom) Normalized integrated peaks against molar ratio (squared symbols) and best fit assuming 1:1 complexation (lines). | ||
Conformational and CD calculations
Out of the four initial positions selected for the docking of each KP enantiomer in β-CyD, we found that three of them result in a deep inclusion of S-KP in the cavity, with comparable energies of 105.8 kcal mol−1 for the B ring entering the secondary rim, 104.5 kcal mol−1 for the A ring entering the primary rim, and 101.2 kcal mol−1 for the B ring entering the primary rim. Analogously, deep inclusion of R-KP was observed for entrance through the primary rim of either B or A rings with energies of 101.9 and 102.1 kcal mol−1, respectively, and through the secondary rim with the B ring (E = 104.7 kcal mol−1). Both enantiomers show unfavourable penetration in the cavity for entrance of the A ring though the secondary rim. The CD spectra of the two diastereomeric complexes show a marked difference of sign in the region of the first electronic transition S1←S0 (n,π*) with a negative signal for S-KP and positive for R-KP, whereas the second transition S2←S0 (ππ*) shows a positive contribution for both of them. The calculated rotational strengths at energy minima geometries, obtained through the cycles of conformational optimization, show that only in few cases the CD is correctly reproduced. In particular it was found that, out of the four different starting configurations, only two give rise to the correct sequence of signs for the S enantiomer, and just one for the R enantiomer. The two structures of the S-KP complex are similar, referring to the entrance of the B and A rings through the secondary and primary rims, respectively. Fig. 3a and 3b show reliable configurations for the complexes of S-KP and R-KP. We observe that in both cases the final position of the mono-substituted ring of KP is close to the center of the CyD primary rim, in good agreement with NMR data (see below).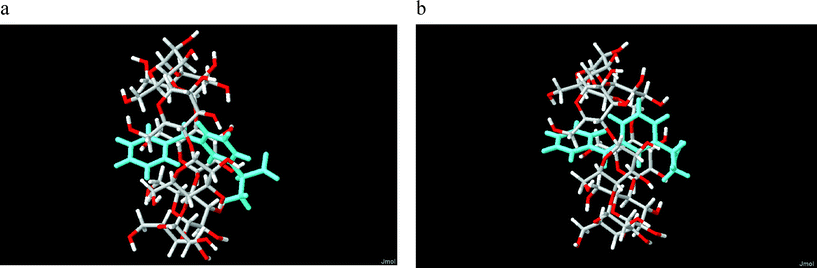 | ||
| Fig. 3 Calculated structures of (a) S-KP and (b) R-KP complexes with β-CyD entering the secondary rim with the B ring. | ||
The calculated rotational strengths for the two most reliable structures found of the S enantiomer complex are represented in Fig. 4a and 5a and for the structure of the R enantiomer complex in Fig. 6a. The individual contributions of the different terms arising from eqn (1) are reported in Fig. 4b, 5b and 6b. It is interesting to notice that in all calculated CD spectra, most of the rotational strength comes out from the second term of eqn (1), i.e. from the interaction of the transition magnetic dipole moment of KP with the electric transition moments of the macrocycle. This term appears particularly important for the nπ* states such as S1, and in fact has been invoked to be determinant to reproduce correctly the intrinsic CD of many proteins, where it arises from excitation of the peptide bond.28 It is satisfying to observe that the sign of the signal relative to the optical activity of the two diastereoisomeric complexes is calculated correctly for both low-lying electronic transitions, despite the approximation of the semi-empirical Hamiltonian used in this work. However, the method tends to overestimate the magnetic terms, resulting in a too large CD signal for the first transition nπ*, with respect to that of the second one of ππ* nature, which appears much larger in the experimental spectrum.
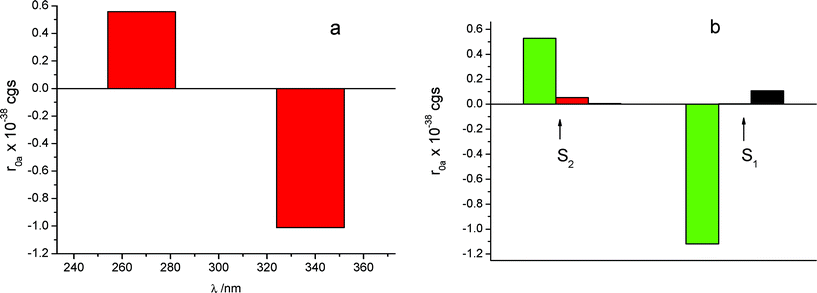 | ||
| Fig. 4 (a) CD calculated for the structure depicted in Fig. 3a, with S-KP entering the secondary rim of β-CyD with the mono-substituted ring, for the S1 and S2 excited states. (b) Calculated contributions deriving from eqn (1): black, intrinsic CD; red, dipole–dipole contribution; green, m-μ mechanism. | ||
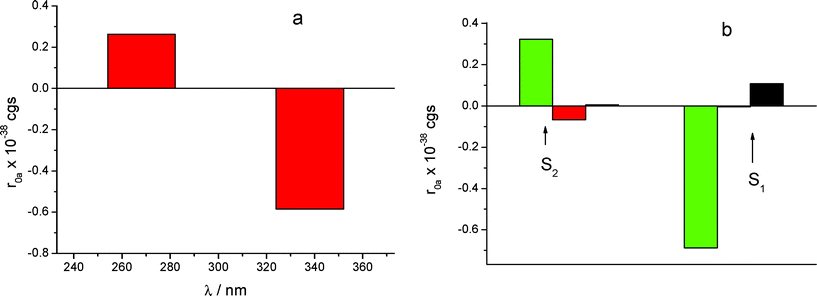 | ||
| Fig. 5 (a) CD calculated for the structure (not displayed), with S-KP entering the primary rim with the di-substituted ring, for the S1 and S2 excited states. (b) Contributions of the various mechanisms. Legend same as in Fig. 4b. | ||
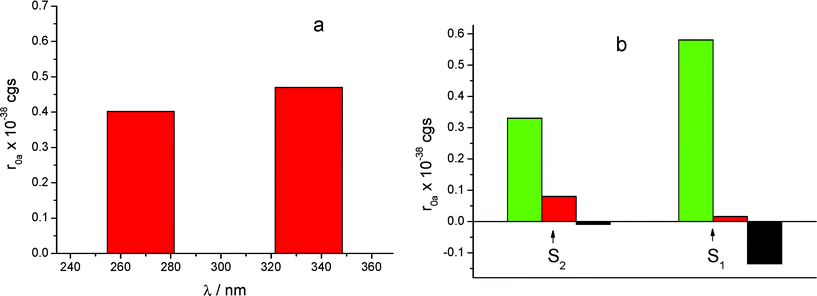 | ||
| Fig. 6 (a) CD calculated for the structure of the R-KP complex displayed in Fig. 3b, for the S1 and S2 excited states. (b) Contributions of the various mechanisms. Legend same as in Fig. 4b. | ||
NMR measurements
1H NMR spectra of the optical antipodes of KP were recorded in 3 × 10−3 M D2O solutions at 25 °C, adjusting pH ≈ 9 by addition of Na2CO3 to the NMR sample in order to ensure complete dissociation of the drug carboxylic group. The signals of KP are separated in distinct spin systems corresponding to the ortho, meta, and paraprotons of both the A and B aromatic rings of the structure reported in Scheme 1. Spectral assignments of all resonances (Fig. 7, spectruma) were achieved by comparison with literature data.35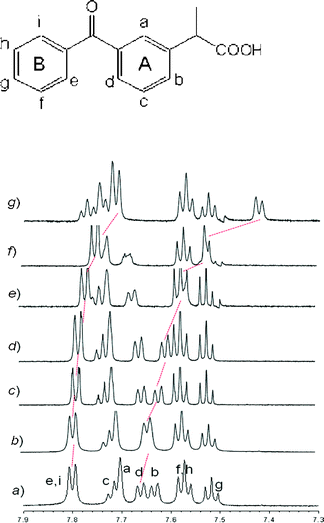 | ||
| Fig. 7 Partial 1H NMR spectra (600 MHz, D2O, 298 K) of aromatic region of a) 3 mM S-KP; and S-KP/β-CyD molar ratio b) 1:0.09; c) 1:0.20; d) 1:0.25; e) 1:0.33; f) 1:0.48; g) 1:1.06. The signals were confirmed also on the basis of 2D ROESY experiments by using the letters reported in the molecular structure. Red dotted lines represent the shift of aromatic protonse and d after addition of β-CyD to the sample solution. | ||
The signals of both enantiomers recorded in the presence of increasing amount of β-CyD showed some differences with respect to those of the free KP. In particular ortho-aromatic protons (labeled with e/i and d) underwent a substantial upfield shift of 0.10 and 0.24 ppm, respectively in a 1:1 KP/β-CyD solution (Fig. 7, line g), this behavior being a clear indication of the occurrence of a “host–guest” interaction.36 In the same equimolar solution the other protons of the B ring of KP gave a minor upfield shift, while the remaining signals of the A ring (except d) together with proton attached to the chiral carbon (δ 3.70 ppm) and methyl group (δ 1.41 ppm) displayed downfield shift upon complexation in the range 0.05–0.08 ppm. Although shielding effects (upfield shifts) are expected due to inclusion, the deshielding phenomena recorded for KP after addition of an equimolar amount of β-CyD (downfield shifts) are interpreted in terms of a possible dissociation of KP dimers. The dimerization phenomena of KP were studied25 by 1H NMR self-titration experiments, carried out in the concentration range 0.5–5.8 mM and the dimer formation was detected by a significant concentration-dependence shielding of all the KP resonances. According to the authors,25 complexation with CyD causes dimer dissociation and overrules the shielding effect caused by inclusion in the CyD cavity. As our NMR spectra were measured for 3mM solutions of KP, dimers might be present and account for the deshielding phenomena recorded for some signals of the guest.
Dimerization of KP is not expected to play any significant role in the conditions of the optical spectroscopic and ITC experiments which were carried out at lower concentrations.
The solution structures of the complexes between β-CyD and R-KP and S-KP were investigated by 1H-1H NOE experiments in order to evaluate the geometry of the associates and to estimate eventual differences of the inclusion direction of the guest or of the position of the chiral center within the cyclodextrin. 2D ROESY spectra were carried out in solutions containing different host/guest stoichiometric ratios (0.09:1; 0.13:1; 0.20:1; 0.25:1; 0.33:1; 0.48:1; 0.67:1; 1.06:1; 1.62:1 for S-KP; 0.08:1; 0.32:1; 1.81:1 for R-KP). It should be pointed out that each enantiomer gives rise to similar NOE interactions. ROESY spectra of water solutions of S-KP containing increasing amounts of β-CyD showed significant interactions of the aromatic protonse/i of A ring and d of B ring of the guest molecule with the inner H3 (medium) and H5 (strong) cyclodextrin protons (Fig. 8). The spectra show also cross-peaks correlating protonsf/h with H5 (medium) as well as with the H6 (medium) protons (data not shown) located at the smaller edge of the macrocyclic host.
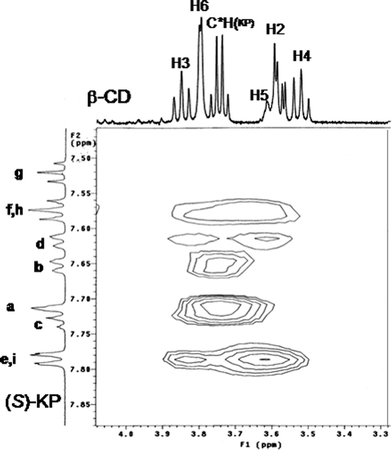 | ||
| Fig. 8 Portion of the ROESY spectrum of a 3 mM solution of S-KP and 0.6 mM of β-CyD in alkaline D2O at 298 K. | ||
Concerning the position of the stereogenic center the interpretation of the NMR spectra of the inclusion complexes was difficult to achieve, due to the overlapping of the cyclodextrin H3-H6 proton signals (3.60–3.90 ppm) with the proton bound to the chiral carbon, while cross peaks between the methyl signal and CD H3, H5, and H6 protons were never observed in each sample investigated. These results suggest that inclusion of S-KP into the CyD cavity occurs from the mono-substituted phenyl portion (B ring) of the drug which locates close to the primary CyD rim and the carboxyl group of the guest exposed to the solvent from the secondary side of the host. As stated above similar results were obtained for the inclusion complex of R-KP. The spectra recorded in alkaline deuterated water at the same concentration of the S enantiomer show the same type of NOE interactions. From the correlation pattern between the B ring and protond of A ring with the inner cyclodextrin protons the geometry of the inclusion can be inferred to be very similar in both the diastereomeric complexes with the B ring of the molecule deeply included inside the CyD cavity.
However, in the study of the complex with the R enantiomer measured in the same host/guest ratio of the S stereoisomer, the presence of an additional cross-peak due to NOE interaction between the aromatic protonse/i (B ring) and H6 CD protons is indicative of a more profound interaction of R-KP with the primary edge of the cavity when compared to the corresponding S enantiomer (Fig. 9).
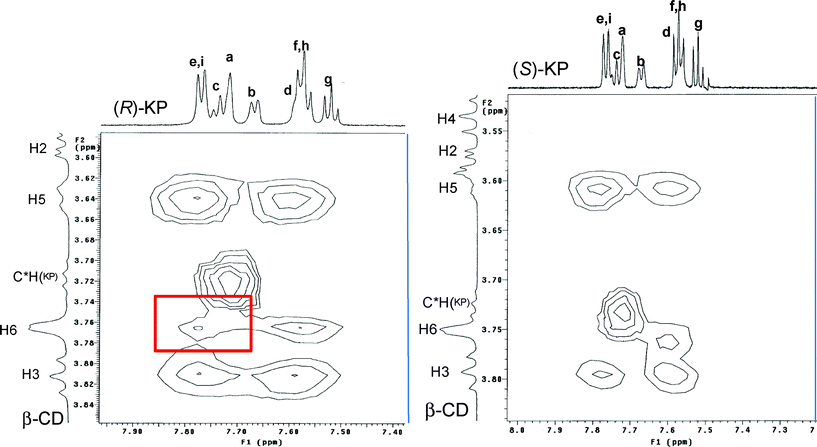 | ||
| Fig. 9 Portion of the ROESY spectra of a 3 mM solution of S-KP (right, X range from 7.20 to 8.02 ppm; Y range from 3.50 to 3.85 ppm) and R-KP (left, X range from 7.38 to 7.96 ppm; Y range from 3.59 to 3.85 ppm) containing 1 mM of β-CyD in alkaline D2O at 298 K. The red rectangle in the spectrum of R enantiomer evidences the cross peak connecting H6 CyD protons with ring B protons (e,i) which is not observed in the case of the S enantiomer. | ||
Photolysis of KP:β-CyD complexes
355 nm laser excitation of a 1 × 10−3 M S-KP or R-KP solution in the presence of β-CyD 1 × 10−2 M in Ar-saturated 0.1 M phosphate buffer of pH 7.4 led to a differential absorption spectra similar for both enantiomers with maxima at λ < 340 nm and λ ∼ 530–540 nm and a broad absorption extending to 700 nm (Fig. 10).37 The ΔA at 340 and 530 nm decay biexponentially with rate constants k1 = (2.0 ± 0.3) × 107 s−1 and k2 = (4.0 ± 0.6) × 106s−1, also similar for both enantiomers. At λ ∼ 600 nm the decay looks like a monoexponential with an intermediate rate constant. After decay of these transients a differential absorption remains, characterized by maxima at λ < 340 nm and ca. 520 nm, disappearing on the microsecond time scale. Similar results had been found with rac-KP complexed to β-CyD in an analogous study carried out with a 35 ps laser pulse at 266 nm for excitation.5,38 According to the previous interpretation the k1 component, dominating at 340 and 530 nm, is assigned to the enantiomeric triplet KP undergoing slow decarboxylation in the β-CyD cavity (k1 ≈ 107s−1 has to be compared to k ∼ 4 × 109 s−1 in water).5,6 The residual 520 nm absorption at 450 ns is attributed to a neutral decarboxylated intermediate converting to the final product 3-ethylbenzophenone (EB, see Scheme 2) on the microsecond/millisecond time scale.5 The k2 component had been previously assigned to a parallel photoreductive path of the KP triplet leading to KP photoaddition to β-CyD. However the structure of the final photoproducts, ascertained in this work and discussed in the following, led us to revise the assignment of this component.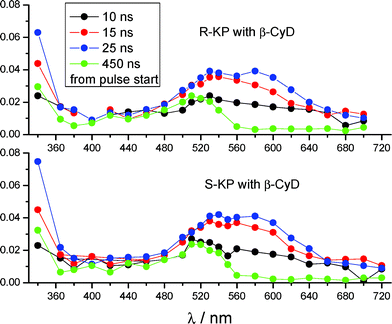 | ||
| Fig. 10 (a) Difference absorption spectra generated after laser excitation at 355 nm of a Ar-saturated solution of 1 × 10−3 M S-KP or R-KP with 0.01 M β-CyD, in 0.1 M phosphate buffer at pH 7.4 at 295 K (3.8 mJ/pulse, optical path 1 cm). | ||
 | ||
| Scheme 2 A proposal for the photochemistry of enantiomeric KP in the β-CyD cavity at neutral pH. | ||
The transient data clearly evidence that the spectral and kinetic evolution of photoexcited KP is not stereodifferentiated in the β-CyD cavity. This reflects also in a lack of stereoselectivity in the photoproducts, as it is shown by the time course of the absorbance at 259 nm (At259) of Ar-saturated KP-β-CyD mixtures irradiated at λ > 305 nm (Fig. 11a), in the early stages of the phototransformation. In principle both photodecarboxylation and H-abstraction from β-CyD could be responsible of the photobleaching of this band, assigned to the lowest π,π* aromatic carbonyl transition. However photodecarboxylation is not expected to actually contribute, because the absorption coefficients of KP and the decarboxylated photoproduct EB, are practically the same at this wavelength. Thus, reasonably, photobleaching of this band mainly occurs by photoreductive destruction of the carbonyl group. The changes in the At259 absorbance are usefully represented by the time dependence of the first derivative dAt259/dt, that appears to be linear with a non-zero slope (negative) and a very small negative intercept (Fig. 11b). These features are consistent with a prevailing role of secondary photochemistry from EB in the photobleaching (reflecting in the non-zero slope), with a very small contribution, if any, from primary KP photochemistry (reflecting in the negative intercept).
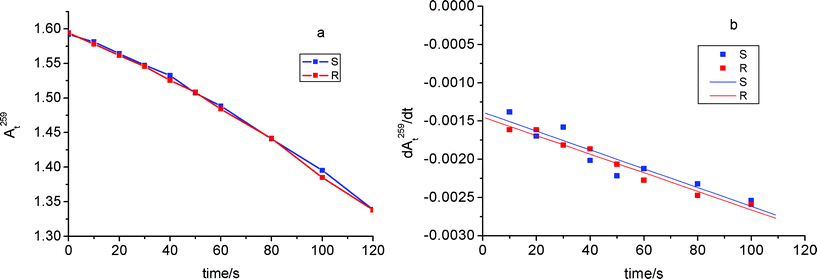 | ||
| Fig. 11 (a) Time course of the absorbance at 259 nm (At259) of Ar-saturated KP-β-CyD mixtures irradiated at λ > 305 nm, in the early stages of the phototransformation: R- and S-KP (1.0 × 10−4M) and β-CyD (1.0 × 10−2 M) in triply distilled water at pH 7.4 adjusted with NaOH. (b) First derivative dAt259/dt. Each point is the average of three independent experiments. Optical path 1 cm. | ||
The mass spectra of the non irradiated KP-β-CyD mixtures (S-KP data in Fig. 12) evidenced the complex at m/z 1387.7 (Fig. 12a). The spectra of the irradiated mixtures showed that EB, with MW 210.0 g mol−1, is the only photoproduct for photobleaching extents <5% (Fig. 12c, peak at m/z 208.9), whereas an additional photoproduct with MW 1344.6 g mol−1 was detected at higher photobleaching percentages (20%) (see peak at m/z 1367.6 corresponding to the Na+ adduct in (+)ESI mass spectrum in Fig. 12d).
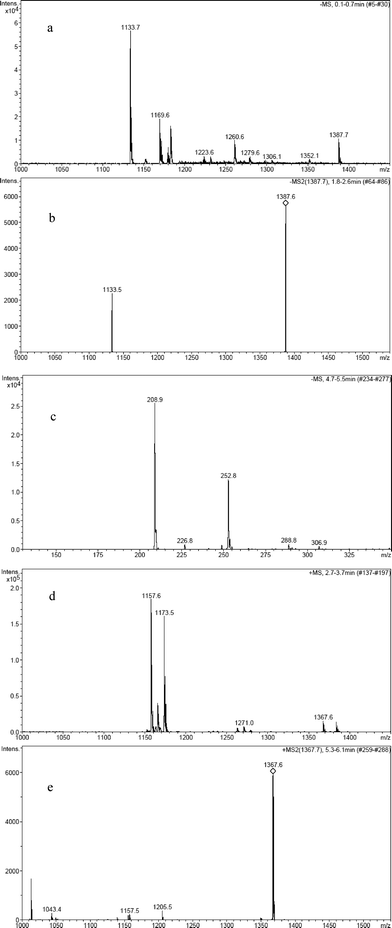 | ||
| Fig. 12 (−)ESI mass spectrum of the non irradiated S-KP-β-CyD complex with peak at 1387.7 (a) and its MS/MS peak fragmentation spectrum (b); (−)ESI mass spectrum of the S-KP-β-CyD complex irradiated under Ar keeping conversion percentage <5% (c); (+)ESI mass spectrum of the S-KP-β-CyD complex irradiated under Ar with conversion percentage of 20% (d) and its MS/MS fragmentation spectrum of the peak with m/z 1367.7 (e). | ||
This molecular weight can in principle correspond to two different species: a non covalent complex of EB with β-CyD or reduced ethylbenzophenone covalently bound to β-CyD. While MS/MS fragmentation of the non irradiated KP:β-CyD complex clearly yielded two peaks attributed to KP and β-CyD (see peak at m/z 1133.5 of β-CyD in Fig. 12b), the MS/MS fragmentation of the photoproduct with MW 1344.6 g mol−1 did not yield β-CyD together with ethylbenzophenone but yielded several peaks (see Fig. 12e for the fragmentation of Na+ adduct). This indicates the product results from the covalent addition of reduced ethylbenzophenone to β-CyD. Appearance of the covalent adduct only after prolonged irradiation clearly indicates it arises from photoreduction of accumulating EB, a very efficient reaction in benzophenone-like derivatives in presence of β-CyD.39,40
The above results led us to reconsider the previous proposal of a photoreductive path for the KP triplet in the β-CyD cavity, in parallel to photodecarboxylation. Indeed because reductive photochemistry pertains to the photodecarboxylated product EB, we assign the k2 component in the kinetic evolution of the transient spectra of Fig. 10 to the carbanion formed upon release of the CO2 fragment from the KP triplet in the β-CyD cavity. This species has λmax = 580 nm6,7 and can be recognized to contribute in part in the difference spectrum taken at 25 ns delay from pulse start, where it is accompanied by the KP triplet with λmax = 520–530 nm. Due to both kinetic and spectral reasons the transients are difficult to disentangle, however the spectral features of the triplet precursor are more evident in earlier difference spectra taken within the laser duration, i.e. at 10–15 ns delay. Decay of the KP carbanion is expected to be controlled by protonation (pKa = 7.6), occurring in aqueous solutions at neutral pH with a rate ∼8 × 106 s−1.6,7,41 A neutral 3-ethylbenzophenone ketyl biradical with λmax = 520 nm, distinguishable in the spectrum at 450 ns delay, was proposed to form.3–6,42 Given the assessed geometry of the KP inclusion (Fig. 3a and 3b) the protonation of the KP carbanion in β-CyD is reasonably not hampered, but the rate of the process (4 × 106s−1) is somewhat lowered with respect to neutral aqueous solution. We propose in Scheme 2 a mechanism for the triplet photochemistry of KP enantiomers in the β-CyD cavity that takes into account most of the above observations.
Conclusions
According to the expectations the chiral recognition ability of β-CyDvs. S- and R-KP enantiomers is rather low. The 1:1 complexes are not characterized by stereodifferentiated stability, the association constants are the same within the experimental uncertainty as determined from both CD and ITC experiments. However the diastereomeric complexes are characterized by well differentiated CD spectra with opposite sign in the lowest energy n,π* band and same sign but different intensity in the second π,π* band. Application of molecular modelling and the quantum mechanical calculation of the induced rotational strengths in the low energy KP:β-CyD associates allowed us to assign them the most likely structure upon comparison of the calculated quantities with the corresponding experimental CD. The geometry of the inclusion is similar for both enantiomers with the aromatic carbonyl inserted in the CyD cavity, the B ring close to the primary CyD rim, the carboxylate group exposed to the solvent from the secondary rim. NMR spectra fully confirmed the geometry of the inclusion of both enantiomers. Tiny structural differences in the diastereomeric complexes were sensibly probed by CD, manifesting in diastereodifferentiated induced signals. Consistently the 2D ROESY spectra for R-KP revealed a cross-peak, due to NOE interaction between aromatic protons of B ring and H6 CyD protons indicative of a closer location of this ring to the primary edge of the β-CyD cavity when compared to the S-enantiomer.
The similarity of the β-CyD inclusion geometry accounts for the lack of chiral discrimination in the photoreactivity of KP within the cavity. Steady state photolysis with UV-absorption and MS detection of photoproducts as well as nanosecond laser flash photolysis evidenced that the only photoreaction of the enantiomeric KP triplet in the β-CyD complex is photodecarboxylation. It was confirmed that release of the CO2 fragment from the carboxylate group, exposed to the solvent from the secondary side of the CyD, occurs with a rate reduced by two orders of magnitude with respect to water, reasonably because the deep insertion of the aromatic carbonyl within the apolar cavity disfavours stabilization of the primary product, the triplet carbanion. MS detection of the final products revealed that the decarboxylated photoproduct, 3-ethylbenzophenone, upon absorption of a second photon, undergoes reductive photochemistry by abstracting H from a CyD glucose unit and eventually leading to a covalent CyD adduct (see Scheme 2).
As concluding remarks we observe that both NMR and CD spectroscopy were able to evidence tiny structural differences in the KP diastereomeric complexes that reflected in the CD spectra and in the NOE interactions, but neither in the thermodynamic binding parameters nor in the photochemistry. Further, the parallel study with NMR and CD allowed to confirm the reliability of the approach based on molecular modelling and rotational strength calculations for the assessment of the structure of low energy guest-CyD associates.
Acknowledgements
This work was supported in part by the EU through the project ITN 237962 CYCLON. The VP-ITC instrument is the property of CIRB (Consorzio Interdipartimentale di Ricerche Biotecnologiche, Universita'di Bologna).References
- F. Bosca, M. L. Marin and M. A. Miranda, Photochem. Photobiol., 2001, 74, 637–655 CAS.
- P. Ghezzi, G. Melillo, C. Meazza, S. Sacco, L. Pellegrini, C. Asti, S. Porzio, A. Marullo, V. Sabbatini, G. Caselli and R. Bertini, J. Pharmacol. Exp. Ther., 1998, 287, 969–974 CAS.
- T. Suzuki, T. Okita, Y. Osanai and T. Ichimura, J. Phys. Chem. B, 2008, 112, 15212–15216 CrossRef CAS.
- C. D. Borsarelli, S. E. Braslavsky, S. Sortino, G. Marconi and S. Monti, Photochem. Photobiol., 2000, 72, 163–171 CrossRef CAS.
- S. Monti, S. Sortino, G. De Guidi and G. Marconi, New J. Chem., 1998, 22, 599–604 RSC.
- S. Monti, S. Sortino, G. De Guidi and G. Marconi, J. Chem. Soc., Faraday Trans., 1997, 93, 2269–2275 RSC.
- L. J. Martinez and J. C. Scaiano, J. Am. Chem. Soc., 1997, 119, 11066–11070 CrossRef.
- S. Monti, I. Manet, F. Manoli, R. Morrone, G. Nicolosi and S. Sortino, Photochem. Photobiol., 2006, 82, 13–19 CAS.
- S. Monti, F. Manoli, S. Sortino, R. Morrone and G. Nicolosi, Phys. Chem. Chem. Phys., 2005, 7, 4002–4008 RSC.
- C. Festa, N. Levi-Minzi and M. Zandomeneghi, Gazz. Chim. Ital., 1996, 126, 599–603 CAS.
- S. Monti, I. Manet, F. Manoli and S. Sortino, Photochem. Photobiol. Sci., 2007, 6, 462–470 RSC.
- S. Monti, S. Ottani, F. Manoli, I. Manet, F. Scagnolari, B. Zambelli and G. Marconi, Phys. Chem. Chem. Phys., 2009, 11, 9104–9113 RSC.
- S. Monti, I. Manet, F. Manoli, S. Ottani and G. Marconi, Photochem. Photobiol. Sci., 2009, 8, 805–813 RSC.
- S. Monti, I. Manet, F. Manoli and G. Marconi, Phys. Chem. Chem. Phys., 2008, 10, 6597–6606 RSC.
- S. Monti, I. Manet, F. Manoli, M. L. Capobianco and G. Marconi, J. Phys. Chem. B, 2008, 112, 5742–5754 CrossRef CAS.
- H. Huhnerfuss and M. R. Shah, J. Chromatogr., A, 2009, 1216, 481–502 CrossRef.
- E. L. Izake, J. Pharm. Sci., 2007, 96, 1659–1676 CrossRef CAS.
- C. J. Easton and S. F. Lincoln, Chem. Soc. Rev., 1996, 25, 163–170 RSC.
- A. M. Abushoffa, M. Fillet, P. Hubert and J. Crommen, J. Chromatogr., A, 2002, 948, 321–329 CrossRef CAS.
- E. Ameyibor and J. T. Stewart, J. Pharm. Biomed. Anal., 1998, 17, 83–88 CrossRef CAS.
- M. Blanco, J. Coello, H. Iturriaga, S. Maspoch and C. Perez-Maseda, J. Chromatogr., A, 1998, 799, 301–307 CrossRef CAS.
- M. Blanco, J. Coello, H. Iturriaga, S. Maspoch and C. Perez-Maseda, J. Chromatogr., A, 1998, 793, 165–175 CrossRef CAS.
- M. Blanco, J. Coello, H. Iturriaga, S. Maspoch and C. Perez-Maseda, Anal. Chim. Acta, 2000, 407, 233–245 CrossRef CAS.
- M. Blanco, J. M. Gonzalez, E. Torras and I. Valverde, Anal. Bioanal. Chem., 2003, 375, 157–163 CAS.
- S. Rozou, S. Michaleas and E. Antoniadou-Vyza, J. Chromatogr., A, 2005, 1087, 86–94 CrossRef CAS.
- G. Marconi, S. Monti, F. Manoli, A. Degli Esposti and B. Mayer, Chem. Phys. Lett., 2004, 383, 566–571 CrossRef CAS.
- G. Marconi, S. Monti, F. Manoli, A. Degli Esposti and A. Guerrini, Helv. Chim. Acta, 2004, 87, 2368–2377 CrossRef CAS.
- P. Bortolus, G. Marconi, S. Monti and B. Mayer, J. Phys. Chem. A, 2002, 106, 1686–1694 CrossRef CAS.
- L. L. Costanzo, G. De Guidi, G. Condorelli, A. Cambria and M. Fama, Photochem. Photobiol., 1989, 50, 359–365 CrossRef CAS.
- InsightII, Accelrys software Inc., San Diego, CA, 2005 Search PubMed.
- W. Chen, C. E. Chang and M. K. Gilson, Biophys. J., 2004, 87, 3035–3049 CrossRef CAS.
- I. J. Tinoco, Adv. Chem. Phys., 1962, 4, 113–161.
- Hyperchem 6.02, Hypercube Inc., Gainesville, FL, 2000 Search PubMed.
- J. H. Obbink and A. M. F. Hezemans, Chem. Phys. Lett., 1977, 50, 133–137 CrossRef CAS.
- E. Skordi, I. D. Wilson, J. C. Lindon and J. K. Nicholson, Xenobiotica, 2004, 34, 1075–1089 CrossRef CAS.
- H. J. Schneider, F. Hacket, V. Rudiger and H. Ikeda, Chem. Rev., 1998, 98, 1755–1785 CrossRef CAS.
- 80–90% of KP is complexed.
- In ref. 5 the rate constant k2 had been assigned to the combination reaction of the radical pair made by the KP ketyl radical and the β-CyD radical, resulting from the H-abstraction from β-CyD by the ketone triplet, to give a covalent KP-β-CyD adduct with λmax = 205 nm.
- S. Monti, N. Camaioni and P. Bortolus, Photochem. Photobiol., 1991, 54, 577–584 CrossRef CAS.
- S. Monti, L. Flamigni, A. Martelli and P. Bortolus, J. Phys. Chem., 1988, 92, 4447–4451 CrossRef CAS.
- G. Cosa, L. Llauger, J. C. Scaiano and M. A. Miranda, Org. Lett., 2002, 4, 3083–3085 CrossRef CAS.
- H. Suzuki, T. Suzuki, T. Ichimura, K. Ikesue and M. Sakai, J. Phys. Chem. B, 2007, 111, 3062–3068 CrossRef CAS.
| This journal is © The Royal Society of Chemistry and Owner Societies 2011 |