Efficient photosensitization of terbium ions enabled by hydrolysis of siloxy groups in ligands with specific side-chains†
Received
20th August 2010
, Accepted 6th October 2010
First published on 26th October 2010
Abstract
We have demonstrated the first observable case of efficient energy transfer from poly-ligands to terbium ions through cross-linked siloxane in solution. The intensification of green luminescence was easily perceived by the naked eye under UV-254 nm illumination, where even the terbium dopants reached 0.1 μM. Moreover, the nano-scale fibrous structure (diameter 100–200 nm) aggregated to a micro-meter size round plate (diameter 1.1 μm). In addition, compound 1 equipped with three long tetradecyl chains in gels gained more of an advantage over the doubly tetradecylated compounds 2 and 3 in terms of energy migration to terbium ions due to side chain effects.
Introduction
Among a rich variety of metal centered functional materials, 4f orbital elements such as lanthanide containing systems are the subject of intensive research due to their fascinating luminescence properties, with high color purity and long lifetimes as well as their potential applications in bioassays and sensors or in a series of light converting devices.1 In order to realize the photonic applications of lanthanide ions, they have usually been introduced into an inert inorganic host. A common problem arising from the above method is the non-homogenous dispersion of the metal ion which is responsible for a reduction in luminescence by the clustering of lanthanide ions. In recent years, another interesting approach concerns the complexation of lanthanides with organic chromophores that are covalently bonded to the hybrid networks, and the corresponding preparation of xerogels or thin films.2 However, the insolubility of the dried gels leads to difficulties in studying the inside structure in detail. The essential characteristics of the above silicate gels are their short-range ability to self-assemble during the sol–gel process. Moreover, metal ions tend to behave differently when placed in confined spaces and the limitation of metal ions in nano or micro-scale enclosures will result in drastic alteration in the physical and chemical properties.3 Therefore, a class of aromatic acids that are suitable for terbium emissions with three or two long alkyl chains to increase the solubility, were modified. The strategy is given schematically in Fig. 1. The rational design is simply to induce terminal triethoxysilane hydrolysis and oligomerize it into a cross-linked surface, and this unusual aggregation of functional groups will bring an unexpected change for terbium sensitization. As estimated, the cross-linked oligomers can be dissolved in CHCl3, CH2Cl2 or THF which we can readily study through NMR, dynamic light scattering (DLS), atomic force microscopy (AFM) and emission spectra. To the best of the authors' knowledge, this is the first case with regard to studying the correlations between organic chromophores bearing hydrolyzed triethoxysilane and terbium ions in solution. Additionally, the specific numbers of aliphatic chains on the ligands are also crucial for sensitizing the central metal ions. To date, relatively few publications have concentrated on this field which described the influence of different linked side-chains on the solution and gel state of the terbium complexes.4
 |
| | Fig. 1 Schematic representation of compounds 1–3 and their hydrolysis reactions. | |
Experimental
General
All the starting materials were obtained from commercial suppliers and used as received. DMF was distilled under reduced pressure and other solvents were used as provided. Visible and fluorescence spectra were measured on Hitachi U-3500 and F-4500 spectrometers respectively. Infrared spectra were measured with a Shimadzu FTIR-8600 spectrometer. Luminescent quantum yields were measured at room temperature using a Hamamatsu photonics absolute PL quantum yield measurement system C9920-02. Lifetime was measured on an Edinburgh FLS920 spectrometer. AFM images were obtained by using a Nanoscope IIIa Tapping mode AFM machine provided by Veeco, Japan. 1H-NMR spectra were recorded with a JEOL AL600 (600 MHz) at 298 K using tetramethylsilane as the internal standard. Dynamic light scattering was measured with a Shimadzu IG-1000. Gel permeation chromatography was investigated with a Viscotek TDAmax. LC-MS spectra were recorded on a Shimadzu LCMS-20/0EV.
The synthetic procedures for the carboxylic acid parts in the amides are available from the literature5,6 and the full data will be reported elsewhere.
Synthesis of compound 16.
3,4,5-Tris(tetradecoxy)benzoic acid (0.4 g) was fully dissolved in CH2Cl2 (15 ml), and HOBt (89.2 mg) and EDC·HCl (122 mg) were added to the mixture. 3-Aminopropyltriethoxysilane (0.236 g) was then added dropwise into the solution slowly; the reaction was monitored by thin layer chromatography, and the amidation was almost finished after 2 h. The mixture was washed with aq. 4% NaHCO3 and aq. sat. NaCl, dried over Na2SO4, and then concentrated in vacuo. Flash column chromatography on silica gel with CHCl3–methanol = 100/1 and then recrystallized from methanol gave analytically pure 1 as a white solid (0.33 g, 65% yield). Mp: 50–52 °C. 1H-NMR (CDCl3) δ = 6.96 (2H, s, 2,6-H), 6.34 (1H, t, J = 6.4 Hz, NH), 3.99 (2H, t, J = 6.4 Hz, 4-OCH2), 3.98 (4H, t, J = 6.0 Hz, 3,5-OCH2), 3.82 (6H, q, J = 6.8 Hz, Si(OCH2)3), 3.44 (2H, q, J = 6.4 Hz, NCH2), 1.80 (8H, m, 3,4,5-OCCH2, NCCH2), 1.46 (6H, m, 3,4,5-OC2CH2), 1.25 (60H, m, 3,4,5-OC3C10H20), 1.22 (9H, t, J = 6.8 Hz, SiO(CCH3)3), 0.89 (9H, t, J = 6.8 Hz, 3,4,5-OC13CH3), 0.70 (2H, t, J = 8.0 Hz, SiCH2). MS (APCI) found m/z 963 (M+), calcd for C58H111NO7Si: 963.
Synthesis of compound 2.
In a similar manner to compound 1, amidation of 3,5-bis(tetradecoxy)benzoic acid gave 2 (70% yield): 1H-NMR (CDCl3) δ = 6.84 (2H, s, 2,6-H), 6.52 (1H, s, 4-H), 6.49 (1H, t, J = 6.4 Hz, NH), 3.93 (4H, t, J = 6.4 Hz, 3,5-OCH2), 3.82 (6H, q, J = 6.4 Hz, Si(OCH2)3), 3.45 (2H, q, J = 6.4 Hz, NCH2), 1.75 (6H, m, 3,5-OCCH2, NCCH2), 1.42 (4H, m, 3,5-OC2CH2), 1.25 (40H, m, 3,5-OC3C10H20), 1.20 (9H, t, J = 6.8 Hz, SiO(CCH3)3), 0.85 (6H, t, J = 6.8 Hz, 3,5-OC13CH3), 0.68 (2H, t, J = 8.0 Hz, SiCH2). MS (APCI) found m/z 750 (M+), calcd for C44H83NO6Si: 750.
Synthesis of compound 3.
In a similar manner to compound 1, amidation of 4-methoxy-3,5-bis(tetradecoxy)benzoic acid gave 3 (61% yield). 1H-NMR (CDCl3) δ = 6.98 (2H, s, 2,6-H), 6.34 (1H, t, J = 6.4 Hz, NH), 3.98 (4H, t, J = 6.4 Hz, 3,5-OCH2), 3.84 (3H, s, OCH3), 3.52 (6H, q, J = 6.0 Hz, Si(OCH2)3), 3.34 (2H, q, J = 6.4 Hz, NCH2), 1.84 (6H, m, 3,5-OCCH2, NCCH2), 1.47 (4H, m, 3,5-OC2CH2), 1.24 (40H, m, 3,5-OC3C10H20), 1.20 (9H, t, J = 7.6 Hz, SiO(CCH3)3), 0.91 (6H, t, J = 6.8 Hz, 3,5-OC13CH3), 0.71 (2H, t, J = 8.0 Hz, SiCH2). MS (APCI) found: m/z 780 (M+), calcd for C45H85NO7Si: 780.
Synthesis of cross-linked compound 17.
6.7 mg of 1 was dissolved in EtOH (0.8 ml) and this was named as solution A. 20 vol% aq. HCl (pH 1.0) in EtOH was prepared and called solution B. Solution B (0.8 ml) was then slowly added into solution A (0.8 ml) with stirring and heated at 50 °C for 6 h. The complete reaction of ethoxysilane groups was shown by the disappearance of the peak at 3.82 ppm in the 1H-NMR. The transparent film like solid was precipitated and filtered by separation. The crude product was washed with ethanol and water three times and dried under vacuum. The product dissolved easily in CHCl3 and THF. Cross linked compounds 2 and 3 were also prepared by the same method.
Results and discussion
NMR investigation
Compound 1 was dissolved in CDCl3 and the ethoxy methylene groups on the silicon atom gave the quartet peak at 3.82 ppm (Figure S1a†). The siloxy moieties were completely reacted and changed to the corresponding silanol with a CD3OD–D2O mixture containing HCl as indicated by the loss of CH2 in the ethoxysilane and the appearance of a signal at 3.61 ppm (CH2 in ethanol, Figure S1b). After polycondensation, compound 1 was not soluble in methanol at all, even at high temperature, and precipitated out; we then collected the solid which was washed with methanol, and dried under vacuum overnight. It was fully dissolved in CDCl3 and Figure S1c shows the NMR spectra. The signals became broadener due to hydrolysis and the broad peak at around 3.95 ppm corresponds to OCH2C13 in the aliphatic chain. The peaks of the Si-OCH2CH3 group at 3.82 and 1.21 ppm had disappeared. Additionally, after gel permeation chromatography (GPC), we found a sole hydrolyzed oligomer in the product, showing that all of compound 1 had disappeared after the acid-catalyzed reaction (Figure S2). To further investigate the molecular weight of the above aggregates, we analyzed its solution in THF by GPC, where the system was calibrated using polystyrene standards. The molecular weight was estimated to be 6807 (PDI = 1.18), indicating that cubic silsesquioxanes might have been constructed because the expected molecular weight of such an octamer prepared from hydrolyzed compound 1 is 6811.
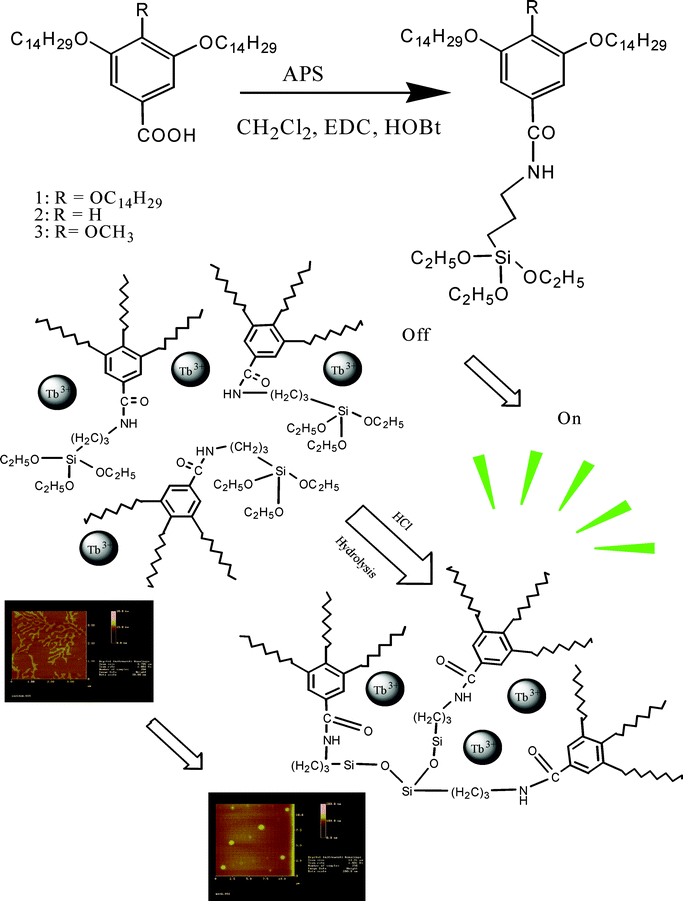
Compound 1 (5 μM) was mixed with a solution of terbium chloride (5 μM) in CHCl3–EtOH = 99/1 (1/Tb = 1/1). As expected, the mixed solution gave very weak luminescence from terbium, showing inefficient energy transfer from compound 1 to terbium ions in a dilute solution (Fig. 2, red line). However, after the acidic hydrolysis and polycondensation processes, a striking fluorescence enhancement was observed with irradiation at 254 nm light (Scheme 1). We can find four characteristic intra-configuration f-f transitions of terbium ions from the 5D4 excited states to the different J levels of the ground term 7FJ (J = 6,5,4,3) in the emission spectra (Fig. 2). The fluorescence intensity of 5D4 → 7F5 located at 547 nm increased almost 80 fold even though the concentration for compound 1 and terbium remained stable. As we decreased the terbium concentration down to 0.1 μM, a distinct green emission could still be observed from the solution. From the spectra, we noticed that the 5D4 → 7F6 and 5D4 → 7F4 transitions included a very weak side band which suggested there may exist two coordination sites for terbium ions in the oligomer solution. In the IR spectra, we already proved that the hydrolytic compound 1 could coordinate to terbium ions and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O located at 1637 cm−1 was shifted to higher frequency to 1645 cm−1 (Δν = 10 cm−1, Figure S3†). Compounds 2 and 3 also show a similar fluorescence turn-on phenomenon after condensation of ethoxysilane groups and the peak intensities of 5D4 → 7F5 are 125 (compound 2) and 110 times (compound 3) that of the non-hydrolyzed molecules respectively (figures are not shown). We measured the quantum yields of the three sets of samples, and the data show that the 10−5 M hydrolyzed sample displayed luminescence of 23 times higher intensity than the non-hydrolyzed one, the same situation also occurred in compounds 2 and 3 (Table 1). Additionally, hydrolyzed samples all exhibited much longer lifetimes in comparison with the intact ones (Table 2).
O located at 1637 cm−1 was shifted to higher frequency to 1645 cm−1 (Δν = 10 cm−1, Figure S3†). Compounds 2 and 3 also show a similar fluorescence turn-on phenomenon after condensation of ethoxysilane groups and the peak intensities of 5D4 → 7F5 are 125 (compound 2) and 110 times (compound 3) that of the non-hydrolyzed molecules respectively (figures are not shown). We measured the quantum yields of the three sets of samples, and the data show that the 10−5 M hydrolyzed sample displayed luminescence of 23 times higher intensity than the non-hydrolyzed one, the same situation also occurred in compounds 2 and 3 (Table 1). Additionally, hydrolyzed samples all exhibited much longer lifetimes in comparison with the intact ones (Table 2).
Table 1 Quantum yields of compounds 1–3 in CHCl3–EtOH = 99/1 with different concentrations (excitation = 285 nm). 1 means free ligand of compound 1 and 1′ means hydrolyzed compound 1
| Concentration/M |
1
|
1
′
|
2
|
2
′
|
3
|
3
′
|
| 10−5 |
0.001 |
0.023 |
— |
0.01 |
— |
0.008 |
| 10−4 |
0.005 |
— |
0.001 |
— |
0.005 |
— |
| 10−3 |
0.015 |
— |
0.002 |
— |
0.008 |
— |
Table 2 Lifetimes of compounds 1–3 in CHCl3–EtOH = 99/1 (10−5M), monitored at terbium emission at 545 nm
| Sample |
1
|
1
′
|
2
|
2
′
|
3
|
3
′
|
| Lifetime/μs |
121 |
972 |
220 |
843 |
182 |
890 |
 |
| | Fig. 2
Emission spectra of compound 1 with Tb ions (1/Tb = 1/1, 5 × 10−6 M, CHCl3–EtOH = 99/1) before (red line) and after hydrolysis (black line). Ex = 280 nm. | |
 |
| | Scheme 1 Compound 1 with terbium ions (1/Tb = 1/1, 5 × 10−6 M, CHCl3–EtOH = 99/1) before (left) and after hydrolysis (right) under UV–254 nm excitation. | |
Generally speaking, the efficiency of the ligand for lanthanide energy transfer requires a compatibility between the ligand excited states and accepting levels of the lanthanide ions, and two energy transfer paths are normally described: the common one is through 1S* (ligand)–3T* (ligand)–4f* and the less frequent one is 1S* (ligand)–4f* direct transfer.8–10 In this case, we investigated the singlet and triplet states of both hydrolyzed and non-hydrolyzed compound 1. From Figure S4,† we can see that the broad band located at 425 nm corresponding to phosphorescence of compound 1 did not shift at all during the hydrolysis reactions. Moreover, the singlet emission bands of the ligands with a maximum at around 320 nm were almost identical with each other, indicating that the energy levels of the two species did not change after cross-linking of the siloxane parts. Additionally, the absorption spectra also provided evidence that compound 1 remained stable because both samples showed overlapping signals with a maximum at 265 nm (Figure S5).
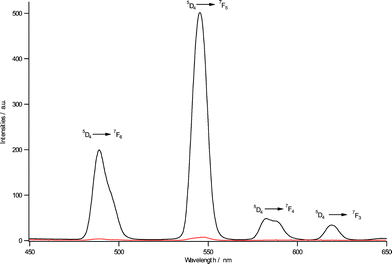
Since the two organic chromophores are very close in their energy levels, the common theory can hardly explain the increase in green emission. The strength of the Ln-ligand interactions and the distance between chelating atoms and lanthanide ions are considered to be decisive factors for this energy transfer process.11 Although it is unrealistic to obtain the real distance from CO to terbium ions, we can investigate the formation of intermolecular aggregation using DLS and AFM during the period of hydrolysis and polycondensation, proving that a high concentration of compound 1, equipped with carbonyl groups, is surrounded with terbium ions, which can dramatically increase the luminescence efficiency. DLS was used to characterize the aggregation of the two samples. The results show that before hydrolysis, compound 1 formed structures with an average diameter of around 200 nm, but after hydrolysis, we noticed the size of aggregates to be significantly increased to 1.1–1.5 μm which is ten times larger than intact molecules (Figure S6†). To study these aggregates further, we applied tapping mode AFM to have a close examination of the two samples (Fig. 3). Clearly, when compound 1 was fully dissolved in CHCl3, it extended to nano-scale fibrous structures with micrometre length and the diameter size ranged from 100–200 nm, which was consistent with its molecular structure based on hydrogen bonds and long alkyl chains. After the hydrolysis reaction, the free ligand of compound 1 aggregated due to the cross-linking of Si–O bonds and the morphology was completely changed. We can find round shaped plates dispersed in CHCl3 solution and obviously the previous fiber structure contracted because of polycondensation. The average diameter of the round plate was about 1.1 μm with the thickness of 26 nm. According to a recent report by Goncalves,12 the condensation of Si–O groups favored branched and cyclic structures rather than linear segments which also coincided with our findings. Compounds 2 and 3 also exhibited similar particle growth and expansion phenomenon with the only difference that the original molecules formed a round shape in chloroform due to decreased solubility (Figure S7). All the AFM results are nearly consistent with the DLS data but with slightly decreased size. It is known to us that the measured AFM height does not exactly correspond to the real distance and is frequently less than the true one in terms of the deformation of the soft surface by the AFM tip. Normally the overall quantum yields of sensitized lanthanide luminescence is given as Φoverall = ΦA·ηET·Φem (ΦA is quantum yield of formation of the intermediate for triplet state; ηET is energy transfer efficiency; Φem emission quantum yields of lanthanide). Clearly, we have substantiated that during the sequence of hydrolysis and polycondensation, ΦA which corresponds to triplet energy levels remained to be almost constant as supported by phosphorescence data. Φem which heavily depends on vibrational energies of the solvents is exactly the same in the reaction. The only variable parameter could be ηET which depends on the distance between the chromophore and terbium ions. In this way, we propose that the hydrolysis and polycondensation reactions made several ligands covalently bonded together through the Si–O network and that these covalent linkages, which are entirely different from common concentrated organic molecules, hold multiple carbonyl groups near to each other and can bind with terbium effectively like a polydentate ligand which will lead to increased stability of terbium ions in solution and allow for the metal center to be free from attack by solvent molecules. Additionally, the hydrolytic compound 1 is somewhat similar to solid xerogel in which the mobility of the chelating species is very limited. The formation of terbium complexes is derived from the close arrangement of the coordination ligands.
 |
| | Fig. 3
AFM of compound 1 (10−3 M, CHCl3) before (left) and after hydrolysis (right). | |
Side chain effects
Although we did not find any difference concerning the energy migration behavior of 1, 2, and 3 in the course of the hydrolysis reaction, it should be noted that from a diluted (10−5 M) to a concentrated chloroform solution (10−3 M) of compounds 1–3, the three compounds exhibit rather different energy migration behavior toward terbium ions. Compound 1 which bears three long aliphatic alkyl chains can partially sensitize terbium ions in 10−5 M but neither 2 nor 3 could do so. At higher concentrations such as 10−3 M, terbium emissions were observed in all the three samples but the quantum yields of 2 and 3 were substantially lower than that of 1 in the gel state (Figure S8,†Table 1).
Because compounds 1–3 were very good organogelators for methanol and ethanol,6 we continued the investigation of terbium luminescence in the gel state (for the comparison with solution samples, the terbium concentration in ethanol gels was fixed at 10−5 M). In the organogel of compound 1, characteristic green emission peaks corresponding to 5D4 → 7F6,5,4 transitions were clearly observed (Fig. 4). In addition, we investigated the temperature dependence of this gel and the green luminescence gradually increased by the heating treatment. However, the organogels of compounds 2 and 3 could not transfer their energy to terbium ions and only emissions from ligands were detected (Fig. 4B).
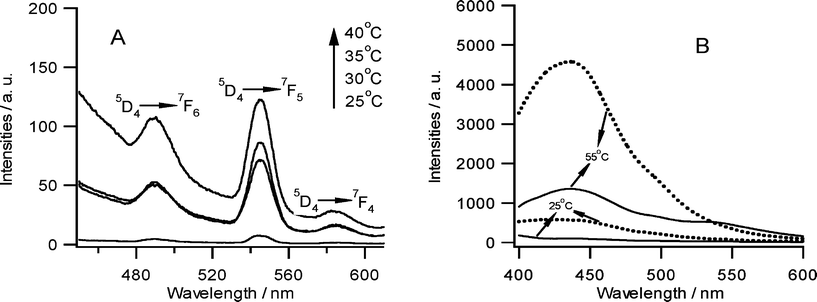 |
| | Fig. 4
A: Emission spectra of the compound 1-ethanol gel doped with terbium ions from 25 to 40 °C (Ex = 317 nm). B: Emission spectra of the compound 2- (dotted line) and compound 3-ethanol gels (solid line) doped with terbium ions at 25 and 55 °C (Ex = 350 nm). Terbium concentration in all the samples was fixed as 10−5 M. | |
Conclusions
We have found a novel and convenient fluorescence turn-on phenomenon concerning the energy transfer to terbium ions with easily accessible triethoxysilane derivatives, through taking advantage of their hydrolysis and polycondensation reactions. At concentrations as low as 10−5 M, the emissions from terbium ions were enhanced by about two orders of magnitude (70, 125, and 110 fold for 1, 2, and 3, respectively) due to the hydrolyzation-induced aggregation. Furthermore, compound 1 with three long aliphatic chains could provide suitable interchain hydrophobic interactions which would protect terbium ions from quenching by solvents or other factors, whereas two chain molecules such as compounds 2 and 3 could not. The presented work gives some alternatives for the future fabrication of structurally controlled lanthanide containing luminescent devices.
Acknowledgements
We thank Drs S. Sasaki, T. Hatano, M. Kunieda and T. Mizoguchi of Ritsumeikan University for useful discussion and Dr T. Hanasaki of Ritsumeikan University for measurements of GPC. Q. Wang appreciates JSPS Post-doctoral Fellowship for Foreign Researchers and the National Natural Science Foundation of China (No.21002035).
References
-
(a) D. Parker, Luminescent lanthanide sensors for pH, pO2 and selected anions, Coord. Chem. Rev., 2000, 205, 109 CrossRef CAS;
(b) C. P. Montgomery, D. Parker and L. Lamarque, Effective and efficient sensitisation of terbium luminescence at 355 nm with cell permeable pyrazoyl-1-azaxanthone macrocyclic complexes, Chem. Commun., 2007, 3841 RSC;
(c) A. S. Chauvin, S. Comby, B. Song, C. D. B. Vandevyver and J. C. G. Bünzli, A versatile ditopic ligand system for sensitizing the luminescence of bimetallic lanthanide bio-imaging probes, Chem.–Eur. J., 2008, 14, 1726 CrossRef;
(d) J. C. G. Bünzli, Benefiting from the unique properties of lanthanide ions, Acc. Chem. Res., 2006, 39, 53 CrossRef;
(e) J. Kido and Y. Okamoto, Organo lanthanide metal complexes for electroluminescent materials, Chem. Rev., 2002, 102, 2357 CrossRef CAS;
(f) B. Viana, N. Koslova, P. Aschehoug and C. Sanchez, Optical-Properties of Neodymium and dysprosium doped hybrid siloxane-oxide coatings, J. Mater. Chem., 1995, 5, 719 RSC;
(g) R. V. Deun, P. Nockemann, C. Gorller-Walrand and K. Binnemans, Strong erbium luminescence in the near-infrared telecommunication window, Chem. Phys. Lett., 2004, 397, 447 CrossRef.
-
(a) A. C. Franville, D. Zambon and R. Mahiou, Luminescence behavior of sol–gel-derived hybrid materials resulting from covalent grafting of a chromophore unit to different organically modified alkoxysilanes, Chem. Mater., 2000, 12, 428 CrossRef CAS;
(b) H. R. Li, J. Lin, H. J. Zhang, L. S. Fu, Q. G. Meng and S. B. Wang, Preparation and luminescence properties of hybrid materials containing europium(III) complexes covalently bonded to a silica matrix, Chem. Mater., 2002, 14, 3651 CrossRef CAS;
(c) Q. M. Wang and B. Yan, Novel luminescent terbium molecular-based hybrids with modified meta-aminobenzoic acid covalently bonded with silica, J. Mater. Chem., 2004, 14, 2450 RSC;
(d) Q. M. Wang and B. Yan, Designing a family of luminescent hybrid materials by 3-(triethoxysilyl)-propyl isocyanate grafted 2-hydroxynicotinic acid bridge molecules, J. Organomet. Chem., 2006, 691, 3567 CrossRef CAS;
(e) Q. M. Wang and B. Yan, Terbium/zinc luminescent hybrid siloxane-oxide materials bridged by novel ureasils linkages, J. Organomet. Chem., 2006, 691, 545 CrossRef CAS;
(f) Q. M. Wang and B. Yan, Optically hybrid lanthanide ions (Eu3+, Tb3+)-centered materials with novel functional di-urea linkages, Appl. Organomet. Chem., 2005, 19, 952 CrossRef CAS;
(g) D. W. Dong, S. C. Jiang, Y. F. Men, X. L. Ji and B. Z. Jiang, Nanostructured hybrid organic-inorganic lanthanide complex films produced in situ via a sol–gel approach, Adv. Mater., 2000, 12, 646 CrossRef.
- R. J. P. Corriu, F. Embert, Y. Guari, C. Reye and R. Guilard, Coordination chemistry in the solid: Evidence for coordination modes within hybrid materials different from those in solution, Chem.–Eur. J., 2002, 8, 5732 CrossRef CAS.
- K. Balakrishnan, A. Datar, T. Naddo, J. L. Huang, R. Oitker, M. Yen, J. C. Zhao and L. Zang, Effect of side-chain substituents on self-assembly of perylene diimide molecules: Morphology control, J. Am. Chem. Soc., 2006, 128, 7390 CrossRef CAS.
- T. Yoshitomi, S. Yabuki, H. Kawakami, R. Sato, K. Toma, M. Furuhata and Y. Maitani, The structure of artificial lipids possessing oligo(ethylene glycol) and their behavior in water, Colloids Surf., A, 2006, 284–285, 276 CrossRef.
- Q. M. Wang, K. Ogawa, K. Toma and H. Tamiaki, A novel luminescent organogel containing dysprosium ions quenched by gel-to-sol transition, Chem. Lett., 2008, 37, 430 CrossRef CAS.
- Y. W. Suh, M. C. Yingmin and H. H. Kung, Size-selective shell cross-linked interior functionalized siloxane nanocages, J. Am. Chem. Soc., 2006, 128, 2776 CrossRef CAS.
- J. C. G. Bünzli, S. Comby, A. S. Chauvin and C. D. B. Vandevyver, New opportunities for lanthanide luminescence, J. Rare Earths, 2007, 25, 257 CrossRef.
- N. S. Baek, Y. H. Kim, S. G. Roh, B. K. Kwak and H. K. Kim, The first inert and photostable encapsulated lanthanide(III) complexes based on dendritic 9,10-diphenylanthracene ligands: Synthesis, strong near-infrared emission enhancement, and photophysical studies, Adv. Funct. Mater., 2006, 16, 1873 CrossRef CAS.
- F. R. G. E. Silva, R. L. Longo, O. L. Malta, C. Piguet and J. C. G. Bünzli, Theoretical modelling of the low quantum yield observed in an Eu(III) triple helical complex with a tridentate aromatic ligand, Phys. Chem. Chem. Phys., 2000, 2, 5400 RSC.
- A. Beeby, D. Parker and J. A. G. Williams, Photochemical investigations of functionalised 1,4,7,10-tetraazacyclododecane ligands incorporating naphthyl chromophores, J. Chem. Soc. Perkin Trans., 1996, 2, 1565 Search PubMed.
- M. C. Goncalves, V. De Zea Bermudez, R. A. Sa Ferreira, L. D. Carlos, D. Ostrovskii and J. Rocha, Optically functional Di-urethanesil nanohybrids containing Eu3+ ions, Chem. Mater., 2004, 16, 2530 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H-NMR, UV-Vis absorption, IR and fluorescence spectra of compound 1. See DOI: 10.1039/c0pp00260g |
| ‡ The present address is School of Chemistry and Environment, South China Normal University, Guangzhou 510006, P. R. China. |
|
| This journal is © The Royal Society of Chemistry and Owner Societies 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.