Highly transparent and luminescent lanthanide ion-containing bridged polysilsesquioxanes
Yige
Wang
* and
Nana
Lin
School of Chemical Engineering and Technology, Hebei University of Technology, Tianjin, 300130, China. E-mail: wangyige@hebut.edu.cn; Fax: +86 225264294; Tel: +86 2260203674
First published on 26th October 2010
Abstract
Novel luminescent bridged polysilsesquioxane monoliths of high transparency have been obtained by hydrolysis and condensation of the precursor under acidic conditions. The monoliths were characterized by FTIR spectroscopy, photoluminescence spectroscopy, and SEM, and they have been used as hosts of Eu3+, Tb3+ and Nd3+ ions. The luminescence lifetime of Tb3+ in this host was 2.1 ms. The interaction between the host and the lanthanide ions could be confirmed by FTIR spectra. The number of water molecules coordinated to the Eu3+ ions was determined to be one. Incorporation of Nd3+ as guest resulted in NIR-luminescent materials.
Introduction
Nanostructured bridged polysilsesquioxanes are a family of hybrid organic-inorganic materials derived from the sol–gel processing of monomers containing a variable organic bridging group and two or more trialkoxysilyl groups.1–3 The bulk properties of such kinds of materials can be tuned by changing the length, rigidity, geometry of substitution, and functionality of the bridging groups that are covalently attached to the trialkoxysilyl groups through Si–C bonds. The main interest in these materials derives basically from the possibility of tailoring the properties of novel multifunctional advanced materials through the combination of the organic and inorganic components at the nanoscaled level in a single material. For instance,4–7dyes have been incorporated into the bridging group of monomers and functional luminescent polysilsesquioxanes have been obtained where a very high loading of dyes without leaching and phase separation can be attained.The burgeoning interest in the investigation on lanthanide (Ln) compounds due to their unique characteristic properties and potential use in a vast range of applications has provided an expanding frontier in materials science.8–10 Although much work has been reported in the incorporation of Ln complexes into hybrid organic-inorganic materials, seeking essentially to take advantage of the high chromaticity and long-lived excited-states characteristic of the metal ions,10–16 less effort has been directed towards the study on the luminescent bridged polysilsesquioxanes containing Ln compounds despite of the significance of combining the sharp emission of Ln ions and the chemically and thermally robust properties of bridged polysilsesquioxanes at molecular levels.17–20
Recently, Carlos and co-workers21–25 have developed a great family of Ln-nanohybrids classified as di-ureasils consisted of urea cross-linking between a silicate backbone and polyether-derived segments of various molecular weights. These materials are full-colour emitters due to the Eu3+ 5D0→7F0–4 red lines overlapped with the broad green-blue emission of the nanoscopic matrices' backbone. Nevertheless, the investigations are mainly focused on Eu3+-doped systems, while Tb3+ and Nd3+-based materials26 are far less involved. Particularly, the study on the highly transparent bridged polysilsesquioxane monoliths containing Tb3+ ions are rarely reported so far. Recent interest in the photophysical properties of Nd3+ ions stems from their possible use in medical diagnostics because the longer emission wavelengths penetrate human tissue far more effectively than does UV or visible light27 In addition, NIR luminescence from Ln3+ ions such as Nd3+ is used as the basis for the optical amplifiers used in fiber optic systems.28
Despite the advances made in the development of bridged polysilsesquioxane, continuous efforts should be made to explore new bis-silylated monomers to obtain advanced polyfunctional organic-inorganic hybrid materials. Herein we report a facile way to prepare a new highly transparent and luminescent bridged polysilsesquioxane monolith incorporating Ln3+ ions rather than Ln complexes by hydrolysis and condensation of novel bis-silylated piperazine-containing monomers (Scheme 1) in the presence of Ln3+ (Ln = Eu, Tb and Nd). The lifetime of Tb3+ excited level of the resulting material is relatively longer (2.1 ms). In addition, the number of water molecules in the first coordination sphere of Eu3+ ions has been calculated on the basis of emission spectrum and lifetime of 5D0 state.
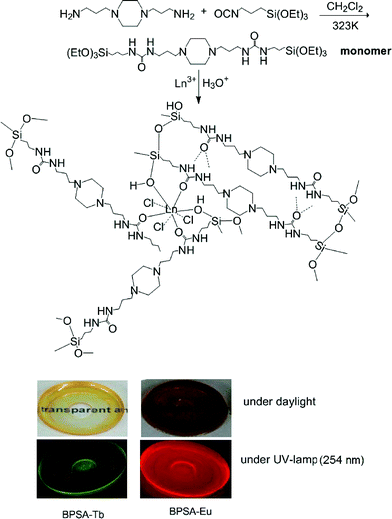 | ||
| Scheme 1 Procedure of preparing luminescent and transparent bridged polysilsesquioxane monoliths. | ||
Results and discussion
All the monoliths are highly transparent and they display green emission for sample BPSA-Tb and red emission for BPSA-Eu (see Scheme 1). Elemental analysis reveals N/C ratios of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 for all samples, consistent with the structure of precursor presented in Scheme 1, indicating that the organic substructure was retained during the hydrolysis–condensation process.
:3 for all samples, consistent with the structure of precursor presented in Scheme 1, indicating that the organic substructure was retained during the hydrolysis–condensation process.
Fig. 1 shows the IR spectra of sample BPSA (a), BPSA-Tb and (b) BPSA-Eu (c). Information concerning hydrogen bonding in the bridged polysilsesquioxanes could be gained from the analysis of the spectral structure in the amide I and amide II spectra regions as highlighted with the red frame. The coordination of the Ln3+ ions to the carbonyl oxygen atoms of the urea linkages could be also assessed in the same region of frequencies.23 The existence of hydrogen bonding between the urea groups in undoped bridged polysilsesquioxanes BPSA is evidenced by IR studies with vibrations at νNH = 3348 cm−1, νCO = 1645 cm−1 and δNH = 1569 cm−1, and these results are similar to those of other bridged polysilsesquioxanes with urea groups.29,30 It has been well established that the difference in the frequencies of maxima of amide I and amide II bands (Δν) can be used to identify the strength of H-bond.31,32 The value of Δ is about 76 cm−1, which indicates the H-bond cannot be characterized as strong.33 Close inspection of the 1850–1450 cm−1 region of the spectra of all the samples depicted in Fig. 1 provides unambiguous evidence that the amide I and amide II bands of the undoped sample are greatly affected by the introduction of guest lanthanide ions. Upon the addition of lanthanide ions, the vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O moieties experiences a dramatic downshift of the 1645 cm−1 to 1622 cm−1 and 1619 cm−1 respectively for Eu3+-doped bridged polysilsesquioxanes (BPSA-Eu) and Tb3+-doped ones (BPSA-Tb.) The interaction between Ln3+ ions and the CO moieties could account for this changes. Furthermore, the amide II mode centered at 1569 cm−1 experiences an up-shift to 1578 cm−1 upon the introduction of lanthanide ions. This means the hydrogen bonds in some of the hydrogen-bonds aggregates formed in the presence of Ln3+ ions are considerably stronger than those in the undoped material. This result is in good agreement with the published data.34 Furthermore, a broad band at 1200–1000 cm−1 ascribing to Si–O–Si asymmetric stretching vibration can be observed in all the spectra and indicates the hydrolysis and condensation of the monomer has occurred.
O moieties experiences a dramatic downshift of the 1645 cm−1 to 1622 cm−1 and 1619 cm−1 respectively for Eu3+-doped bridged polysilsesquioxanes (BPSA-Eu) and Tb3+-doped ones (BPSA-Tb.) The interaction between Ln3+ ions and the CO moieties could account for this changes. Furthermore, the amide II mode centered at 1569 cm−1 experiences an up-shift to 1578 cm−1 upon the introduction of lanthanide ions. This means the hydrogen bonds in some of the hydrogen-bonds aggregates formed in the presence of Ln3+ ions are considerably stronger than those in the undoped material. This result is in good agreement with the published data.34 Furthermore, a broad band at 1200–1000 cm−1 ascribing to Si–O–Si asymmetric stretching vibration can be observed in all the spectra and indicates the hydrolysis and condensation of the monomer has occurred.
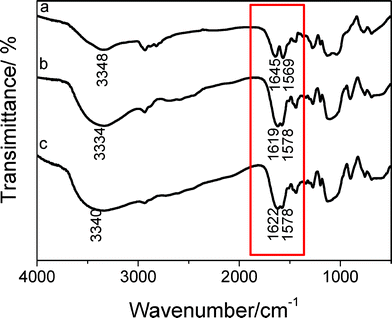 | ||
| Fig. 1 FTIR spectra of material BPSA (a), BPSA-Tb (b) and BPSA-Eu (c). | ||
Fig. 2 shows the scanning electronic microscopy (SEM) images of sample BPSA and BPSA-Eu. The SEM image of sample BPSA shows dense, compact solids with granular structure, whereas the SEM image of BPSA-Eu material shows particle morphology (ca. 0.3 μm) with various sizes and shapes. Similar morphology of Tb-doped material can also be observed (here not shown). The morphological difference between sample BPSA and BPSA-Eu (or BPSA-Tb) could be ascribed to the coordination of Eu3+ to the carbonyl oxygen atoms of the urea linkages during the sol–gel process. The introduction of Ln3+ induces a more positive charge over the urea NH, which thus promotes the formation of additional hydrogen bonds with neighbouring urea groups.24
 | ||
| Fig. 2 SEM images of materials BPSA (top) and BPSA-Eu (down); the scale bar is 2 μm. | ||
The excitation and emission spectra of the Eu3+-doped sample BPSA-Eu are shown in Fig. 3. The excitation spectrum was obtained by monitoring the emission of Eu3+ ions at 612 nm. The spectrum presents a broad band ranging from 250 nm to 400 nm. This band superimposes a series of sharp lines ascribed to an intra-4f6 line (at 320 (7F0→5H6), 365 (7F0 → 5D4), 395 (7F0 → 5L6), 415 (7F0 → 5D3) and 465 nm (7F0 → 5D2).34 The relatively strong intensity of the broad band with respect to the intra-4f6 lines indicates that the luminescence via the excitation of host material is much more efficient than the direct excitation of the Eu3+ absorption levels. The main large broad band might be ascribed to the absorption of the BPSA matrices as evidenced by the spectral overlap between the excitation spectrum of BPSA-Eu and the absorption spectrum of BPSA (Fig. 1s). The sharp lines can be assigned to intra-4f6 transitions. Fig. 3b shows the emission spectrum detected under different excitation wavelengths. The spectra are essentially composed of the Eu3+ typical intra-4f6 lines ascribed to the 5D0 → 7F0–4 transitions. The typical red colour of europium emission is mostly attributed to the strongest transition (5D0 → 7F2) centered at 613 nm. Furthermore, the Eu3+ emission lines are independent of the excitation wavelength within the broad excitation band. Moreover, the detection of a single line for the 5D0 → 7F0 transition, the local-field splitting of the 7F1 level and the higher intensity of the 5D0 → 7F2 transition indicate that the Eu3+ local environment has low symmetry without an inversion centre. The decay curve of 5D0 level is well-reproduced by means of a single-exponential function, the lifetime value is independent of the excitation wavelength and is determined to be 569 ± 2.18 μs (here not shown). The value of lifetime of Eu3+ ions here is shorter than that in the triazine-containing polysilsesquioxanes (τ = 0.744 ms).35
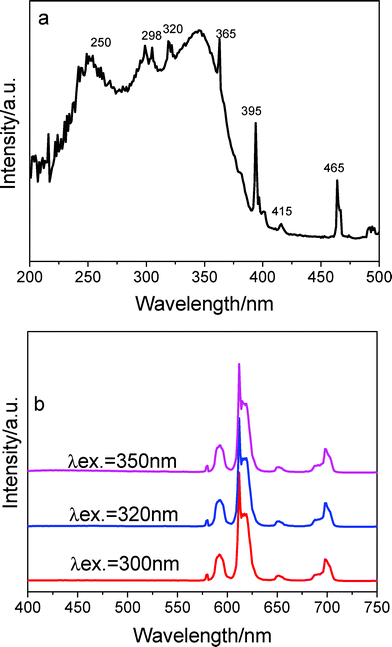 | ||
| Fig. 3 Excitation (a) and emission spectra (b) of sample BPSA-Eu. | ||
The 5D0 quantum efficiency (q) and the number of the water molecules, nw, coordinated to the Eu3+ ion can be estimated on the basis of emission spectrum and lifetime of 5D0 state by using the following equations according to ref. 18. Assuming that only nonradiative and radiative processes are involved in the depopulation of the 5D0 state, q may be defined as
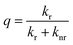 | (1) |
 | (2) |
 | (3) |
| Sample | τ/ms | k r/ms−1 | k nr/ms−1 | q (%) | n w |
|---|---|---|---|---|---|
| BPSA-Eu | 0.569 | 0.233 | 1.52 | 13 | 1.3 |
The luminescence features of Tb3+ doped material (BPSA-Tb) are presented in Fig. 4a and 4b. The excitation spectrum monitored with the emission of Tb3+ ions at 544 nm exclusively consists of a broad band ranging from 220 to 400 nm. Similarly, this band is due to the matrix absorption. Fig. 4b shows the emission spectrum detected under different excitation wavelengths. The spectra present four emissions lines of Tb3+ ions, peaking at 489(5D4 → 7F6), 545 (5D4 → 7F5), 586(5D4 → 7F4) and 620 nm (5D4 → 7F3), with the 5D4→7F5 green emission as the most prominent peak, The luminescence decay of 5D0 level can be well fitted by one exponential. The fitted value for the lifetime is 2102.7 ± 5.8 μs, which is much longer than in other sol–gel-derived organic-inorganic hybrid material.39–41 Although the reason is unclear, it makes sense to assume that reduced phonon relaxation and/or reduced quenching process could be account for this longer lifetime.42–44
 | ||
| Fig. 4 Excitation (a) and emission spectra (b) of sample BPSA-Tb. | ||
The Eu3+ ions can be replaced in this system by other Ln3+ ions that show luminescence in near-infrared regions (Nd). The emission spectrum of the hybrid material BPSA-Nd obtained by excitation at 350 nm is shown in Fig. 5. As shown in Fig. 5, the emission spectrum consists of three sharp bands centered at 883, 1067, and 1344 nm, which are attributed to the f–f transitions of 4F3/2(emitting level)→4I9/2, 4F3/2→4I11/2 and 4F3/2→4I13/2, respectively. The relative intensity sequence of these three transitions is I(4F3/2→4I11/2) > I(4F3/2→4I9/2) > I(4F3/2→4I13/2). Among the three bands of the emission spectra, the intensity of the transition at 1067 nm is the strongest, and for a long time this center has been found to have potential application in laser systems.28 The 1344 nm emission bands of the material offer the opportunity to develop new materials suitable for the optical amplifiers operating at 1.3 μm, one of the telecommunication windows.45
 | ||
| Fig. 5 Emission spectrum of the hybrid material BPSA-Nd. | ||
Conclusions
In conclusion, novel luminescent bridged polysilsesquioxane monoliths with high transparency have been obtained. The highly transparent bridged polysilsesquioxane exhibited characteristic red emission and green emissions upon the addition of Eu3+ ions and Tb3+ ions, respectively. In addition, the obtained material can show luminescence in the near-infrared regions when Nd3+ ions were incorporated with instead. A very long lifetime (2102.7 μs) of Tb3+ ions hosted in bridged polysilsesquioxane monolith has been obtained. The new organic-inorganic hybrid materials are suited to cooperation with UV emitting LEDs because they can be excited in the near UV region. Furthermore, the Nd-containing material has potential application in laser systems and is suitable for the optical amplifiers operating at 1.3 μm.Experimental
Materials
1,4-(3-Aminopropyl) piperazine (Aldrich) and 3-(triethoxysiyl)propyl isocyanate (Aldrich) were used as received. EuCl3·6H2O and TbCl3·7H2O were obtained by dissolving Eu2O3 and Tb4O7 (Tianjin Institute of Fine Chemicals) in HCl solution (37%).Materials preparation
The undoped bridged polysilsesquioxane (BPSA) was prepared as follows: 1,4-(3-aminopropyl) piperazine (1 mmol) was dissolved in 2 mL of dichloromethane solution under a nitrogen atmosphere. 3-(Triethoxysiyl)propyl isocyanate (2 mmol) was slowly added at 50 °C. The mixture was stirred for 5 h and a light yellow solution was developed, 5 mL of ethanol and 0.5 mL of HCl (0.2 M) were then added dropwise. After 30 min, the volume of solvent was evaporated to be about 30% of original size. Then the solution was poured into a plastic box with flat-bottom and was dried at 50 °C for 3 days. The transparent and crack-free monolith was obtained; the lanthanide-containing bridged polysilsesquioxane (BPSA-Eu, BPSA-Tb and BPSA-Nd) were prepared similarly, except that 0.5 mmol of LnCl3 was added.Characterization
Infrared (FTIR) spectra were obtained on a Bruker Vector 22 spectrometer using KBr pellets for solid samples, from 400–4000 cm−1 at a resolution of 4 cm−1 (16 scans collected), about 2 mg of each compound was mixed with potassium bromide (Merck, spectroscopic grade) finely ground and pressed into pellets. Elemental analysis was performed on Elementar Vario EI. SEM images were obtained from an FE-SEM (Hitachi S-4300) at an acceleration voltage of 10 kV. The samples were ground to powder before SEM measurement. The steady-state luminescence spectra and the lifetime measurements were measured on an Edinburgh Instruments FS920P near-infrared spectrometer, with a 450 W xenon lamp as the steady-state excitation source, a double excitation monochromator (1800 lines mm−1), an emission monochromator (600 lines mm−1), a semiconductor cooled Hamamatsu RMP928 photomultiplier tube, and a liquid nitrogen cooled Hamamatsu R5509-72 near infrared photomultiplier tube.Acknowledgements
This work is financially supported by the National Natural Science Foundation of China (No. 20871040, No. 20901022), Program for New Century Excellent Talents in University (NCET-09-0113), Tianjin Natural Science Foundation (09JCYBJC05700), the Key Project of Chinese Ministry of Education (208016), the Scientific Program of Hebei Province (08965110D) and Hebei Province Natural Science Foundation for Distinguished Young Scholars (No. B2010000034)References
- K. J. Shea and D. A. Loy, Acc. Chem. Res., 2001, 34, 707–716 CrossRef CAS.
- R. J. P. Corriu, Angew. Chem., Int. Ed., 2000, 39, 1376–1398 CrossRef CAS.
- J. J. E. Moreau, L. Vellutini, M. Wong, Chi Man, C. Bied, P. Dieudonn, J. L. Bantignies and J. L. Sauvajol, Chem.–Eur. J., 2005, 11, 1527–1537 CrossRef CAS.
- H. S. Peng, J. Tang, J. B. Pang, D. Y. Chen, L. Yang, H. S. Ashbaugh, C. J. Brinker, Z. Z. Yang and Y. F. Lu, J. Am. Chem. Soc., 2005, 127, 12782–12783 CrossRef CAS.
- L. H. Zhao, D. A. Loy and K. J. Shea, J. Am. Chem. Soc., 2006, 128, 14250–14251 CrossRef CAS.
- N. G. Liu, K. Yu, B. Smarsly, D. R. Dunphy, Y. B. Jiang and C. J. Brinker, J. Am. Chem. Soc., 2002, 124, 14540–14541 CrossRef CAS.
- L. D. Carlos, R. A. Sá Ferreira, S. S. Nobre, M. C. Man, J. J. E. Moreau, C. Bied and B. Pichon, Mater. Sci. Forum, 2006, 118, 514–516.
- T. Posati, F. Bellezza, A. Cipiciani, F. Costantino, M. Nocchetti, L. Tarpanni and L. Latternini, Cryst. Growth Des., 2010, 10, 2847–2850 CrossRef CAS.
- J.-C. G. Buenzli and C. Piguet, Chem. Rev., 2002, 102, 1897–1928 CrossRef.
- H. R. Li, J. Lin, H. J. Zhang, L. S. Fu, Q. G. Meng and S. B. Wang, Chem. Mater., 2002, 14, 3651–3655 CrossRef CAS.
- P. Lenaerts, A. Storms, J. Mullens, J. D'Haen', C. Görller-Walrand, K. Binnemans and K. Driesen, Chem. Mater., 2005, 17, 5194–5201 CrossRef CAS.
- L. N. Sun, H. J. Zhang, C. Y. Peng, J. B. Yu, Q. G. Meng, L. S. Fu, F. Y. Liu and X. M. Guo, J. Phys. Chem. B, 2006, 110, 7249–7258 CrossRef CAS.
- L. Armelao, G. Bottaro, S. Quici, M. Cavazzini, M. C. Raffo, F. Barigelletti and G. Accorsi, Chem. Commun., 2007, 2911–2913 RSC.
- E. De Oliveira, C. R. Neri, O. A. Serra and A. G. S. Prado, Chem. Mater., 2007, 19, 5437–5442 CrossRef CAS.
- A. C. Franville, D. Zambon and R. Mahiou, Chem. Mater., 2000, 12, 428–435 CrossRef CAS.
- P. Escribano, B. Julián-López, J. Planelles-Aragó, E. Cordocillo, B. Viana and C. Sanchez, J. Mater. Chem., 2008, 18, 23–40 RSC.
- H. R. Li, J. Lin, H. J. Zhang, H. C. Li, L. S. Fu, Q. G. Meng and S. B. Wang, Chem. Commun., 2001, 1212–1213 RSC.
- M. Fernandes, V. de Zea Bermudez, R. A. Sá Ferreira, L. D. Carlos, A. Charas, J. Morgado, M. M. Silva and M. J. Smith, Chem. Mater., 2007, 19, 3892–3901 CrossRef CAS.
- R. J. P. Corriu, F. Embert, Y. Guari, C. Reye and R. Guilard, Chem.–Eur. J., 2002, 8, 5732–5741 CrossRef CAS.
- L. Raehm, A. Mehdi, C. Wickleder, C. Reye and R. J. P. Corriu, J. Am. Chem. Soc., 2007, 129, 12636–12637 CrossRef CAS.
- R. A. Sá Ferreira, L. D. Carlos, R. R. Gonçalves, J. L. S. Ribeiro and V. de Zea Bermudez, Chem. Mater., 2001, 13, 2991–2998 CrossRef CAS.
- L. D. Carlos, Y. Messaddeq, H. F. Brito, R. A. Sá Ferreira, V. de Zea Bermudez and J. L. Ribeiro, Adv. Mater., 2000, 12, 594–598 CrossRef CAS.
- V. de Zea Bermudez, R. A. Sá Ferreira, L. D. Carlos, C. Molina, K. Dahmouche and S. J. Ribeiro, J. Phys. Chem. B, 2001, 105, 3378–3386 CrossRef.
- S. S. Nobre, X. Cattoen, R. A. Sá Ferreira, C. Carcel, V. de Zea Bermudez, M. Wong, Chi Man and L. D. Carlos, Chem. Mater., 2010, 22, 3599–3609 CrossRef CAS.
- S. S. Nobre, C. D. S. Brites, R. A. Sá Ferreira, V. de Zea Bermudez, C. Carcel, J. J. E. Moreau, J. Rocha, M. Wong, Chi Man and L. D. Carlos, J. Mater. Chem., 2008, 18, 4172–4182 RSC.
- M. C. Gonçalves, N. J. O. Silva, V. de Zea Bermudez, R. A. Sá Ferreira, L. D. Carlos, K. Dahmouche, C. V. Santilli, D. Ostrovskii, I. C. Correia, Vilela and A. F. Craievich, J. Phys. Chem. B, 2005, 109, 20093–20104 CrossRef CAS.
- G. M. Davies, R. J. Aarons, G. R. Motson, J. C. Jeffery, H. Adams, S. Faulkner and M. D. Ward, Dalton Trans., 2004, 1136–1144 RSC.
- L. N. Sun, H. J. Zhang, L. S. Fu, F. Y. Liu, Q. G. Meng, C. Y. Peng and J. B. Yu, Adv. Funct. Mater., 2005, 15, 1041–1048 CrossRef CAS.
- J. J. E. Moreau, L. Vellutini, M. Wong, Chi Man and C. Bied, J. Am. Chem. Soc., 2001, 123, 1509–1510 CrossRef CAS.
- J. J. E. Moreau, B. P. Pichon, M. Wong, Chi Man, C. Bied, H. Pritzkow, J.-L. Bantignies, P. Dieudonne' and J.-L. Sauvajol, Angew. Chem., Int. Ed., 2004, 43, 203–206 CrossRef CAS.
- J. van Esch, F. Schoonbeck, M. de Loos, H. Kooijman, L. A. Spek, R. M. Kellogg and B. L. Feringa, Chem.–Eur. J., 1999, 5, 937–950 CrossRef CAS.
- D. P. Fasce, R. J. J. Williams, L. Matějka, J. Pleštil, J. Brus, B. Serrano, J. C. Cabanelas and J. Baselga, Macromolecules, 2006, 39, 3794–3801 CrossRef CAS.
- J. J. E. Moreau, B. P. Pichon, C. Bied, M. Wong and Chi Man, J. Mater. Chem., 2005, 15, 3929–3936 RSC.
- P. P. Yang, Z. W. Quan, C. X. Li, J. Yang, H. Wang, X. M. Liu and J. Lin, J. Solid State Chem., 2008, 181, 1943–1949 CrossRef CAS.
- N. N. Lin, H. R. Li, Y. G. Wang, Y. Feng, D. S. Qin, Q. Y. Gan and S. D. Qin, Eur. J. Inorg. Chem., 2008, 4781–4785 CrossRef CAS.
- R. M. Supkowski and W. De Horrocks Jr., Inorg. Chim. Acta, 2002, 340, 44–48 CrossRef CAS.
- Y. Feng, H. R. Li, Q. Y. Gan, Y. G. Wang, B. Y. Liu and H. J. Zhang, J. Mater. Chem., 2010, 20, 972–975 RSC.
- K. Binnemans, P. Lenaerts, K. Driesen and C. Goerller-Walrand, J. Mater. Chem., 2004, 14, 191–195 RSC.
- D. W. Dong, S. C. Jiang, Y. F. Men, X. L. Ji and B. Z. Jiang, Adv. Mater., 2000, 12, 646–649 CrossRef.
- F. Y. Liu, L. S. Fu, J. Wang, Q. G. Meng, H. R. Li, J. F. Guo and H. J. Zhang, New J. Chem., 2003, 27, 233–235 RSC.
- B. H. Tong, S. J. Wang, J. Jiao, F. R. Ling, Y. Z. Meng and B. Wang, J. Photochem. Photobiol., A, 2007, 191, 74–79 CrossRef CAS.
- V. Bekiari, P. Lianos and P. Judeinstein, Chem. Phys. Lett., 1999, 307, 310–316 CrossRef CAS.
- D. M. Boye, A. J. Silversmith, T. Nguyen, Nguyen and K. R. Hoffman, J. Non-Cryst. Solids, 2007, 353, 2350–2354 CrossRef CAS.
- T. Ishizaka, R. Nozaki and Y. Kurokawa, J. Phys. Chem. Solids, 2002, 63, 613–617 CrossRef CAS.
- S. I. Klink, P. O. Alink, L. Grave, F. G. A. Peters, J. W. Hofstraat, F. Geurts and F. C. J. M. Van Veggel, J. Chem. Soc., Perkin Trans. 2, 2001, 363 RSC.
| This journal is © The Royal Society of Chemistry and Owner Societies 2011 |