Activation energies of photoinduced unimolecular, bimolecular and termolecular processes on silica gel surfaces
Siân L.
Williams
*,
David R.
Worrall
,
Iain
Kirkpatrick
,
Anisoara
Vancea
and
Jiawei
Pan
Department of Chemistry, Loughborough University, Loughborough, Leicestershire, UK LE11 3TU. E-mail: s.l.williams@lboro.ac.uk
First published on 26th October 2010
Abstract
Activation energies for energy and electron transfer have been measured in various systems on silica gel. In the case of ion-electron recombination, a facile technique involving fluorescence recovery is described which complements diffuse reflectance spectroscopy in the study of these systems. In bimolecular anthracene/azulene systems, activation energies have been shown to be independent of pre-treatment temperature in the range 25–210 °C, demonstrating that physisorbed water plays little role in determining diffusion rates on silica gel. In a ternary anthracene/azulene/perylene system, we have for the first time presented comparative activation energies for the diffusion of azulene and its radical cation, and have shown a greater activation energy for diffusion of the latter species.
Introduction
The photochemistry and photophysics of molecules adsorbed on oxide surfaces,1–25 contained within zeolites25–36 and porous clays,25,37–40 has been the subject of a number of studies. Silica gel is widely used in industry as a catalytic support and a greater understanding of reaction kinetics on these materials would be invaluable, particularly in regards to optimising these applications.The excitation and multi-photon ionisation of arenes and their subsequent energy and electron transfer reactions on silica gel have been previously reported1–4,6–8 including investigation of the influence of the nature and loading of the donor and acceptor compounds on the reaction. The radical cation decays on short timescales (of the order of a few milliseconds) via geminate recombination6 and at longer timescales by diffusion of the radical cation and electron followed by ion-electron recombination. The decay of excited state and radical ion species on silica gel is not uncomplicated due to the heterogeneity of the surface and rates rarely conform to simple exponential kinetics. The model described by Albery et al.41 has been used to characterise the data sets, ensuring a comprehensive exploration of the parameter space in order to obtain a global optimum value for the rate parameters.2,3
Previous studies have shown evidence for energy and electron transfer reactions in these systems as being largely dependent upon the rates of diffusion,2,3 and in the case of anthracene/azulene systems, rates of reaction are governed by the rate of diffusion of azulene which is more mobile than the anthracene moeities. Energetics were also demonstrated to have a significant role in determining electron transfer rates,2,6 and we have shown a Marcus-type dependence of rate on the free energy for electron transfer.2,6 In these studies we have also found some evidence for an additional influence from steric factors.2
In previous work the activation energy for back electron transfer from the surface to the radical cations of anthracene derivatives was reported.1,4 There was a correlation between the activation energy and the size of the radical cation which suggested that movement of the radical cation and not of the electron dominated the observed kinetics.
Fluorescence has been extensively used to study singlet state decay and aggregation effects on various surfaces, including silica gel15,17,20 and cellulose.17 However, the study of the kinetics of decay or reactions of “dark” states such as triplets and radical ions has been through the use of diffuse reflectance spectroscopy.1–3,5–8,12,16–18,21,23,26 This latter technique has been used by ourselves to study the long time decay (greater than 30 s) of anthracene radical cations.1 This technique works well as most anthracene derivatives have a distinctive radical cation absorption peak in the 700–800 nm region.4 In this paper fluorescence is also being used as a complementary technique to study the rate of ion-electron recombination of radical cations of anthracene derivatives. The presence of the radical cation reduces the concentration of ground state anthracene which in turn reduces the amount of fluorescence from the sample. As the sample undergoes ion-electron recombination regenerating the molecule's ground state the observed fluorescence intensity increases as the radical cation decays, and this increase in fluorescence intensity correlates directly with increased ground state population.
The rate of back electron transfer of 2,3-didecyloxyanthracene and 2,3-dimethoxyanthracene radical cations adsorbed on silica gel has been studied by monitoring the fluorescence of the anthracene derivative. As the radical cation diffuses across the surface and recombines with an electron it returns to its ground state thus the fluorescence reappears slowly. Kinetics were studied as a function of temperature and concentration, and activation energies calculated. The activation energies obtained via the fluorescence technique agree well with previous data1 obtained via traditional diffuse reflectance measurements, and indeed has corrected some anomalous points in that reported data. Hence in this paper we report a complimentary technique to diffuse reflectance spectroscopy which is not subject to the same interferences, and therefore can provide useful additional data with which to characterise the systems described. These results reiterate the fact that activation energies for ion-electron recombination on silica gel vary with the identity of the radical cation adsorbed on the surface, thus showing that the diffusion of the radical cation across the surface must contribute to the rate of ion-electron recombination.
In addition we report the activation energy for the diffusion of the azulene radical cation, which we recently showed unambiguously to undergo rapid motion on the silica gel surface,8 and demonstrate that the activation energy is larger than for the neutral molecule.
Experimental
Samples were prepared as follows: silica gel was dried under vacuum to 5 × 10−5 mbar for 8 h at 125 °C and then re-pressurised with dry nitrogen. For the pre-treatment temperature study, drying temperatures ranged from 25 °C to 210 °C. The compound(s) of interest were dissolved in n-hexane or acetonitrile and a known weight added to the dried silica gel. The mixture was allowed to equilibrate for a period of one hour, with periodic agitation. The solvent was removed under vacuum to a pressure of 5 × 10−5 mbar and the sample sealed under vacuum into a cylindrical glass cuvette (22 mm diameter × 10 mm pathlength). Sample loadings were calculated on the basis of the mass and concentration of solution added to the silica gel. Sample concentrations for the 2,3-DMO and 2,3-DDO ranged between 0.5 μmol g−1 and 17 μmol g−1. Sample concentrations for energy and electron transfer were kept constant with an anthracene/2-methylanthracene loading of 1.0 μmol g−1 and an azulene loading of 1.0 μmol g−1. Sample concentrations for electron transfer in the termolecular system were as follows 9-carboxylic acid anthracene 1.0 μmol g−1, azulene 0.5 μmol g−1 and perylene 0.5 μmol g−1. Sample concentrations for all systems studied were kept low corresponding to a homogeneous 2D surface concentration of less than 1% of a monolayer as discussed in previous publications.1–5The following chemicals were used as supplied: silica gel (Davisil grade 635, 60–100 mesh, 6 nm pore size, surface area 480 m2 g−1; Aldrich Chemical Co.), acetonitrile (spectrophotometric grade; Aldrich Chemical Co.), n-hexane (spectrophotometric grade; Aldrich Chemical Co.), anthracene (scintillation grade, minimum 99%, Sigma), 2-methylanthracene (HPLC grade; ≥ 99%; Fluka), 9-anthracenecarboxylic acid (minimum 99%, Aldrich Chemical Co.), azulene (99%; Aldrich Chemical Co.), perylene (99%; Aldrich Chemical Co.), 2,3-didecyloxyanthracene (2,3-DDO) and 2,3-dimethoxyanthracene (2,3-DMO) were kindly supplied by Professors Bouas-Laurent and Desvergne of the University of Bordeaux and prepared as described in the literature.42
Laser excitation for production of the radical cation in the unimolecular reaction (ion-electron recombination) was achieved using the third harmonic of a Surelite I Nd:YAG laser (Continuum). The decay of the radical cation was monitored via fluorescence measurements using a Spex FluoroMAX spectrofluorimeter. The fluorescence was measured using front surface geometry. For varying the temperature the sample was thermostatted in an aluminium sample block with a sapphire window, heated electrically using a heating pad (RS components, 12 V, 2 W). Fluorescence spectra were measured using an excitation wavelength of 375 nm with the monitoring wavelength being chosen as the fluorescence maximum observed in the emission spectrum. The fluorescence recovery was measured every 20 s over a 2 h period.
Flash photolysis experiments were used for studying electron and energy transfer in the bimolecular and termolecular systems. The nanosecond diffuse reflectance laser flash photolysis apparatus has been described previously9,10 and excitation of the samples was with the third harmonic (355 nm) of a Surelite I Nd:YAG laser (Continuum). The pulse energy was attenuated using solutions of sodium nitrite in water, to ensure that transient reflectance changes were kept below 10%, where the change in reflectance is directly proportional to the concentration of transient species.43–45 Diffusely reflected analysing light from a 300 W xenon arc lamp (Oriel) was collected and focused onto the entrance slit of an f/3.4 grating monochromator (Applied Photophysics) and detected with a side-on photomultiplier tube (Hamamatsu R928). Signal capture was achieved by using an LT364 Waverunner digitising oscilloscope (LeCroy), interfaced to an IBM-compatible PC.
Activation energies for triplet–triplet energy transfer and electron transfer were measured by heating the samples using a custom built aluminium heating block and a thermal heater pad (JR Tech; 50 mm diameter, 30 V, 15 W). Transients were recorded for triplet state and radical cation decay over the temperature range 25–75 °C, during both heating and subsequent cooling of the block in order to minimise effects due to thermal lag between block and sample cell.
Transient decay data has been analysed using the model of Albery et al. described previously2,3,41 according to eqn (1)
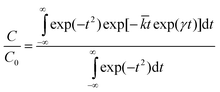 | (1) |
![[k with combining macron]](https://www.rsc.org/images/entities/i_char_006b_0304.gif) is the mean rate constant. The method for fitting transient decays using this model has been described previously in references 1 and 2. At low sample loadings and small reflectance changes (less than 10%), C and C0 can be replaced by ΔR and ΔR0 (or F and F0), the reflectance changes (or fluorescence intensity change) at times t = t and t = 0 (relative to the laser pulse) respectively. This model has been used extensively for data characterisation in these systems previously, and shown to characterise the data with a well-defined minimum in the reduced χ2 parameter space.3 Other models have been applied including the use of a Lévy flight model2,46–48 and the use of Excel's Solver49 where more complex systems are concerned, but the addition of such extra parameters was not found necessary in this data.
is the mean rate constant. The method for fitting transient decays using this model has been described previously in references 1 and 2. At low sample loadings and small reflectance changes (less than 10%), C and C0 can be replaced by ΔR and ΔR0 (or F and F0), the reflectance changes (or fluorescence intensity change) at times t = t and t = 0 (relative to the laser pulse) respectively. This model has been used extensively for data characterisation in these systems previously, and shown to characterise the data with a well-defined minimum in the reduced χ2 parameter space.3 Other models have been applied including the use of a Lévy flight model2,46–48 and the use of Excel's Solver49 where more complex systems are concerned, but the addition of such extra parameters was not found necessary in this data.
Results and discussion
Activation energies have been measured for a number of systems on silica gel; ion-electron recombination in a unimolecular system, both triplet–triplet energy transfer and electron transfer in a bimolecular system, and electron transfer in a termolecular system.Activation energies for ion-electron recombination
Rate constants for the ion-electron recombination of 2,3-DMO and 2,3-DDO radical cations were measured as a function of temperature. Both diffuse reflectance and fluorescence recovery techniques were used, the results from the diffuse reflectance having been previously reported.1 The fluorescence spectra of 2,3-didecyloxyanthracene (adsorbed on silica gel) at various time delays following laser excitation are shown in Fig. 1. Laser excitation produces the radical cation thus causing a reduction in fluorescence intensity, which over time returns to or near its initial intensity. The fluorescence peak has a maximum around 410 nm for 2,3-DDO and the 2,3-DMO spectrum is similar with its peak at 405 nm, thus these wavelengths were chosen for monitoring the fluorescence recovery for these samples. As found previously,1 the 2,3-DDO radical cations recombine with ejected electrons more slowly than the corresponding 2,3-DMO samples of comparable concentrations. The fluorescence spectra are entirely superimposable throughout the radical cation decay, as illustrated in the inset of Fig. 1 for 2,3-DMO. The fact that the spectra are superimposable and return to approximately the initial intensity suggests that no fluorescent products are formed and that the ground state is regenerated, to within error, quantitatively as the radical cation decays. However the identical experiment conducted using unsubstituted anthracene shows that successive normalised fluorescence spectra are not superimposable and the overall fluorescence intensity does not return to the initial value. Fig. 2 shows the fluorescence spectra of anthracene adsorbed on silica gel following laser excitation. These spectra have been normalised to demonstrate that in addition to the initial drop in intensity and slow recovery of fluorescence following laser excitation, the fluorescence spectra change in shape. The shorter wavelength peak decreases slightly relative to the others and the longer wavelength peaks increase during the ion-electron recombination process, illustrative of the formation of a fluorescent product. As a consequence, fluorescence cannot therefore reliably be used to monitor the decay of the anthracene radical cation in this instance, as recovery of ground state fluorescence would need to be deconvolved from the growth of fluorescence of the product, which in all probability will have a different fluorescence quantum yield. Hence in order for this technique to be used reliably, it must first be established that fluorescent products are not formed, i.e. that the normalised fluorescence spectra are superimposable. In practice this is found to be the case for many anthracene derivatives, not all of which are reported here. These observations are consistent with the work of Dabestani et al., who observed photolysis products from anthracene on silica gel, but not from 2-substituted anthracenes.23,50 They ascribed this effect to the lack of formation of stable ground-state pairs in the case of substituted anthracenes.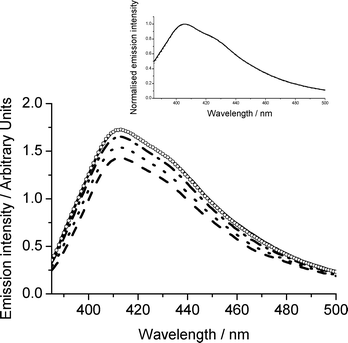 | ||
Fig. 1 Fluorescence recovery of 2,3-DDO on silica gel (3.5 μmol g−1, temperature 316 K) as a function of time following laser excitation (355 nm). Before irradiation (![[thick line, graph caption]](https://www.rsc.org/images/entities/char_e117.gif) ), 30 s after ( ), 30 s after (![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) ), 3 min after (⋯), 15 min after ( ), 3 min after (⋯), 15 min after (![[dash dot, graph caption]](https://www.rsc.org/images/entities/char_e090.gif) ), 60 min after (○). Inset: Normalised fluorescence recovery for 2,3-DMO on silica gel (3.1 μmol g−1, temperature 316 K) as a function of time following laser excitation (355 nm). ), 60 min after (○). Inset: Normalised fluorescence recovery for 2,3-DMO on silica gel (3.1 μmol g−1, temperature 316 K) as a function of time following laser excitation (355 nm). | ||
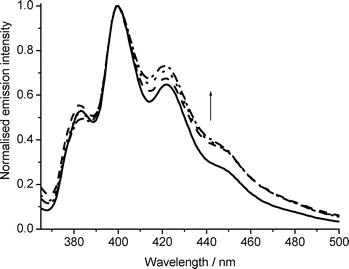 | ||
| Fig. 2 Normalised fluorescence recovery of anthracene on silica gel (1 μmolg−1, temperature 316 K) as a function of time following laser excitation (355 nm). Before irradiation (), 1 min after (), 14 min after (⋯), 66 min after (). | ||
It should be noted that this observation is not limited to this technique; where a fluorescent or absorbing product has a spectrum overlapping significantly with the species of interest, similar complications arise irrespective of the technique employed. Anthracene ion-electron recombination can be reliably studied using diffuse reflectance provided any products formed do not absorb at 715 nm.
The kinetics of fluorescence recovery for two particular anthracene derivatives, 2,3-DMO and 2,3-DDO, were studied as a function of temperature. This enabled the activation energy for the ion-electron recombination process to be determined using Arrhenius plots. Fig. 3 shows a plot of ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) kversus 1/T for 2,3-DMO (1.1 μmol g−1), with k determined from fluorescence recovery, which yields a slope of Ea/R. The activation energy values obtained and previously reported1 using both diffuse reflectance spectroscopy and the fluorescence recovery technique are shown in Table 1 for comparison. As can be seen there is excellent agreement between the parameters extracted from the two techniques. In fact, data obtained from fluorescence recovery “corrects” anomalous points extracted from diffuse reflectance spectroscopy (see Fig. 4). This illustrates the utility of this technique as complimentary to diffuse reflectance spectroscopy in studying kinetics in such systems. As observed previously,1 the 2,3-DDO samples yield much larger activation energies than the equivalent 2,3-DMO samples. The activation energies for both of the derivatives decrease with increasing concentration, as shown in Table 1. The activation energies obtained for 2,3-DMO using the fluorescence recovery and diffuse reflectance techniques are shown together for comparison in Fig. 4. The fluorescence recovery data for 2,3-DMO agrees very well with the diffuse reflectance data, as illustrated in Fig. 4 and Table 1.
kversus 1/T for 2,3-DMO (1.1 μmol g−1), with k determined from fluorescence recovery, which yields a slope of Ea/R. The activation energy values obtained and previously reported1 using both diffuse reflectance spectroscopy and the fluorescence recovery technique are shown in Table 1 for comparison. As can be seen there is excellent agreement between the parameters extracted from the two techniques. In fact, data obtained from fluorescence recovery “corrects” anomalous points extracted from diffuse reflectance spectroscopy (see Fig. 4). This illustrates the utility of this technique as complimentary to diffuse reflectance spectroscopy in studying kinetics in such systems. As observed previously,1 the 2,3-DDO samples yield much larger activation energies than the equivalent 2,3-DMO samples. The activation energies for both of the derivatives decrease with increasing concentration, as shown in Table 1. The activation energies obtained for 2,3-DMO using the fluorescence recovery and diffuse reflectance techniques are shown together for comparison in Fig. 4. The fluorescence recovery data for 2,3-DMO agrees very well with the diffuse reflectance data, as illustrated in Fig. 4 and Table 1.
| System | Concentration/μmol g−1 | Diffuse reflectance/kJ mol−1 | Fluorescence recovery/kJ mol−1 |
|---|---|---|---|
| 2,3-DMO | 0.46 | 53 ± 2 | 54 ± 4 |
| 1.1 | 35 ± 2 | 51 ± 2 | |
| 3.1 | 40 ± 1 | 44 ± 2 | |
| 5.6 | 28 ± 2 | 28 ± 1 | |
| 2,3-DDO | 0.54 | 77 ± 10 | — |
| 1.5 | 68 ± 5 | 70 ± 5 | |
| 3.5 | 30 ± 4 | 58 ± 7 | |
| 16.6 | 48 ± 2 | — |
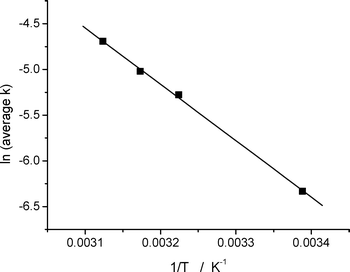 | ||
| Fig. 3 Arrhenius plot for the ion-electron recombination of 2,3-DMO on silica gel (1.1 μmol g−1) following laser excitation (355 nm). | ||
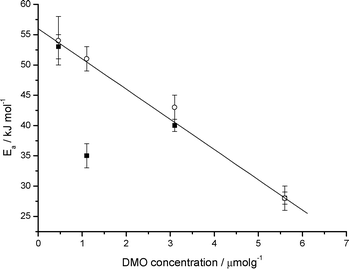 | ||
| Fig. 4 Activation energies for ion-electron recombination of 2,3-DMO radical cations on silica gel as a function of 2,3-DMO concentration measured using diffuse reflectance (■) and fluorescence recovery (○). The error bars shown represent the precision of the measurement. | ||
A similar trend is observed for the 2,3-DDO data, the activation energies again showing a decrease with increasing concentration. This trend in activation energy is rationalised1 on the basis that the “stronger” adsorption sites on silica gel are occupied first and as the concentration increases these sites are filled leaving only the “weaker” adsorption sites at higher concentrations. Those molecules adsorbed at “stronger” sites will be more tightly bound and thus have higher activation energies for movement than those at the weaker sites. Hence the observed activation energy will decrease with increasing concentration as more and more molecules will occupy the weaker sites as the number of molecules increases. This hypothesis is supported by inspection of the gamma values (a measure of the deviation from mono-exponential behaviour) associated with the measured decay rate constants. In the case of 2,3-DMO gamma increases from 1.5 to 2.9 as loading increases; however the trend in the case of 2,3-DDO is less obvious.
Activation energies in bimolecular systems
Rate constants for the decay of anthracene triplet state and radical cation in the presence of 1.0 μmol g−1azulene have been measured. Activation energies for energy and electron transfer in this bimolecular system have also been measured as a function of pre-treatment temperature (PTT) of the silica gel. Quenching constants and activation energies for both electron and energy transfer were measured, but no noticeable dependence on pre-treatment temperature was observed. An Arrhenius plot for electron and energy transfer for the 2-methylanthracene/azulene system is shown in Fig. 5. The effect of pre-treatment temperature on the diffusion of molecules adsorbed on silica gel has been studied previously21,22 but over larger temperature ranges (from 25 up to 800 °C) with conflicting results. Bjarenson and Peterson22 showed that the diffusion coefficients of tetracene on silica with 150 Å pores remained approximately constant when the pre-treatment temperature was increased from 25 to 600 °C, but that the mobile fraction increases with increasing temperature. However, Oelkrug et al.21 reported a reduction in the mobility of aromatic nitrogen heterocycles on silica gel having 60 Å pores with increasing pre-treatment temperature of the surface.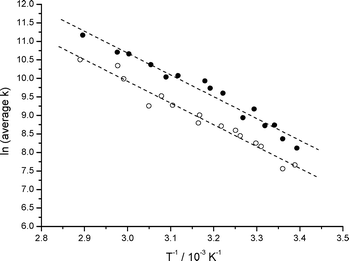 | ||
| Fig. 5 Arrhenius plots for energy transfer (●) and electron transfer (○) between 2-methylanthracene (1.0 μmol g−1) and co-adsorbed azulene (1.0 μmol g−1) on silica gel, PTT 95 °C, following laser excitation at 355 nm. | ||
Table 2 summarises the results obtained and it can be seen that to within experimental error there is little or no dependence of activation energy (Ea) on the pre-treatment temperature (PTT) or the identity of the sensitiser. Electron transfer from azulene to the anthracene/2-methylanthracene radical cation and triplet energy transfer from anthracene/2-methylanthracene to azulene yield the same activation energies. The similarity of the results is not surprising as in both cases the azulene is diffusing across the silica surface followed by either electron transfer to the anthracene radical cation or energy transfer from the anthracene triplet. These values are similar to those reported for diffusion of anthracenes in a polymer matrix51 and slightly higher than the values reported for the quenching of the duroquinone triplet with triphenylamine on silica gel.52
| Species | PTT/°C | E a/kJ mol−1 | A/1012s−1 |
|---|---|---|---|
| An T1 | 25 | 49 ± 3 | 1.2 |
| An •+ | 25 | 50 ± 2 | 7.3 |
| An T1 | 105 | 48 ± 2 | 0.7 |
| An •+ | 105 | 52 ± 2 | 3.1 |
| An T1 | 205 | 48 ± 3 | 1.6 |
| An •+ | 205 | 51 ± 2 | 3.2 |
| 2-MeAn T1 | 95 | 49 ± 3 | 2.1 |
| 2-MeAn •+ | 95 | 49 ± 2 | 0.9 |
| 2-MeAn T1 | 210 | 53 ± 3 | 6.7 |
| 2-MeAn •+ | 210 | 52 ± 5 | 1.5 |
This suggests that the energy barrier for the energy and electron transfer process as observed is independent of the amount of adsorbed water, since based on previous studies53 there should be significant desorption of physisorbed water over the temperature range studied here. On the timescales over which these reactions occur, which are of the order of milliseconds, the lifetimes of the excited triplet state and radical cation are significantly enhanced relative to their counterparts in solution. At the low sample loadings employed in this study, the presence of surface water might be expected to present an impediment to diffusion and reduce the chance of an encounter with an energy acceptor/electron donor molecule. In addition, it may be expected that physisorbed water would block certain adsorption sites. This former effect is confirmed using Monte-Carlo simulations, the details of which have been published elsewhere.1 However it is clear from the results presented here that physisorbed water does not present an obvious impediment to surface diffusion at these low substrate loadings, nor does physisorbed water significantly alter the adsorption of the substrates.
Activation energies in termolecular systems
In a previous publication,8 we described a termolecular system which for the first time unambiguously demonstrated radical cation diffusion on silica gel, and indeed showed that this diffusion rate in the case of the azulene radical cation was approximately a factor of 10 slower than that of the neutral molecule. Here we extend this study to encompass activation energies, since the activation energy for the diffusion of an uncharged species can be compared with that of its radical cation through the study of either the decay of the primary electron acceptor or the rise of the tertiary donor. The system chosen was 9-anthracenecarboxylic acid (9CAn)/azulene (Az)/perylene (Pe). The 9CAn was chosen because it is strongly adsorbed to the silica surface, and hence does not contribute to the observed diffusional kinetics. Following laser excitation the 9CAn is photoionised to its radical cation. Azulene then transfers an electron to the 9CAn radical cation, regenerating the 9CAn ground state and forming the Az radical cation which then diffuses across the surface. When it encounters a Pe ground state (which is diffusing relatively much more slowly across the silica gel surface8), the redox potentials2,54 are such that Pe transfers an electron to the Az reforming the Az ground state and generating the Pe radical cation. A kinetic trace showing the decay of the 9CAn radical cation and the rise of the Pe radical cation is shown in Fig. 6. Consequently monitoring the rate of decay of the 9CAn radical cation as a function of temperature allows determination of the activation energy for the diffusion of the azulene ground state, whilst monitoring the rise of the Pe radical cation as a function of temperature allows determination of the activation energy for the diffusion of the azulene radical cation.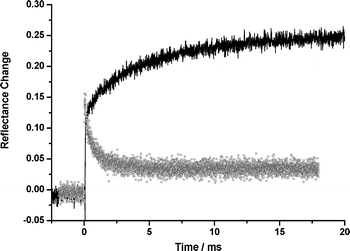 | ||
| Fig. 6 Decay of the 9CAn radical cation (○) monitored at 715 nm and rise of the Pe radical cation () monitored at 530 nm with Az adsorbed as a hole shuttle on silica gel following laser excitation (355 nm). | ||
As shown in Table 3, the activation energy for the quenching of the anthracene radical cation by azulene is independent of whether anthracene or 9-anthracenecarboxylic acid is employed as the electron acceptor, demonstrating that the kinetics are dominated by the rate of azulene diffusion. The rise of the perylene radical cation in the termolecular system has an activation energy about 10 kJ mol−1 higher than that for the anthracene decay, reflecting the higher activation energy for diffusion of the radical cation. This is to our knowledge the first comparison of activation energies for diffusion of a neutral molecule and its radical cation on silica gel.
| System | Species | E a/kJ mol−1 | A/1012s−1 |
|---|---|---|---|
| AnAz | An•+ decay | 52 ± 2 | 3.1 |
| 9CAnAzPe | 9CAn•+ decay | 51 ± 1 | 3.1 |
| 9CAnAzPe | Pe•+ rise | 63 ± 3 | 4.1 |
Conclusions
In this paper we have demonstrated that over the pre-treatment temperature range 25–210 °C, activation energies for electron and energy transfer do not change to within the experimental error. Activation energies obtained for ion-electron recombination on silica gel have been measured both using the diffuse reflectance method and the more accessible fluorescence recovery method. The values obtained have been shown to be independent of monitoring method, thus proving fluorescence recovery as a useful technique for monitoring back electron transfer on surfaces. A facile technique for determining whether fluorescence recovery may reliably be used in a particular system is described. Using a termolecular electron transfer system we have for the first time independently determined activation energies for the diffusion of a neutral molecule and its radical cation on silica gel, and have demonstrated that the activation energy for diffusion of the radical cation is higher by around 10 kJ mol−1 for this particular system.Acknowledgements
The authors wish to thank Professor Henri Bouas-Laurent and Professor J.-P. Desvergne for their gift of the derivatised anthracenes used in this work. We would also like to thank EPSRC for funding this project.References
- D. R. Worrall, S. L. Williams and T. Ganguly, Ion-electron recombination on silica gel surfaces: experiment and modelling, Photochem. Photobiol. Sci., 2006, 5, 844–849 RSC.
- D. R. Worrall, I. Kirkpatrick and S. L. Williams, Bimolecular processes on silica gel surfaces: energetic factors in determining electron-transfer rates, Photochem. Photobiol. Sci., 2004, 3, 63–70 RSC.
- D. R. Worrall, I. Kirkpatrick and S. L. Williams, Controlling factors in electron and energy transfer reactions on silica gel surfaces, Photochem. Photobiol. Sci., 2002, 1, 896–901 RSC.
- D. R. Worrall, S. L. Williams, F. Wilkinson, J. E. Crossley, H. Bouas-Laurent and J. P. Desvergne, Spectroscopy and ion-electron recombination kinetics of radical ions of anthracenes and substituted anilines on silica gel, J. Phys. Chem. B, 1999, 103, 9255–9261 CrossRef CAS.
- D. R. Worrall, S. L. Williams and F. Wilkinson, Electron transfer on insulator surfaces: Exciplex emission and the role of electron diffusion in determining radical deactivation rates, J. Phys. Chem. A, 1998, 102, 5484–5490 CrossRef CAS.
- D. R. Worrall, S. L. Williams and F. Wilkinson, Electron transfer reactions of anthracene adsorbed on silica gel, J. Phys. Chem. B, 1997, 101, 4709–4716 CrossRef CAS.
- F. Wilkinson, D. R. Worrall and S. L. Williams, Primary Photochemical Processes of Anthracene Adsorbed on Silica-Gel, J. Phys. Chem., 1995, 99, 6689–6696 CrossRef CAS.
- S. L. Williams, I. Kirkpatrick and D. R. Worrall, Electron transfer reactions in ternary systems on silica gel surfaces: evidence for radical cation diffusion, Photochem. Photobiol. Sci., 2010, 9, 937–941 RSC.
- D. R. Worrall and S. L. Williams, Diffuse-Reflectance Laser Flash Photolysis, Encyclopedia of Modern Optics Volume 1, R. D. Guenther, D. G. Steel and L. Bayvel (eds), Elsevier, Oxford, 2004, pp. 31–38 Search PubMed.
- F. Wilkinson and R. Beer, Diffuse reflectance laser photolysis of adsorbed molecules, in Photochemical processes in organised molecular systems, K. Honda, Editor, 1991, Elsevier Science, Amsterdam, pp. 377–398 Search PubMed.
- J. P. Da Silva, E. V. Bastos, L. V. F. Ferreira and R. G. Weiss, Surface photochemistry of the herbicide napropamide. The role of the media and environmental factors in directing the fates of intermediates, Photochem. Photobiol. Sci., 2008, 7, 69–75 RSC.
- L. F. V. Ferreira, J. P. Da Silva, I. F. Machadoa, T. J. F. Branco and J. C. Moreira, Surface photochemistry: Dibenzo-p-dioxin adsorbed onto silicalite, cellulose and silica, J. Photochem. Photobiol., A, 2007, 186, 254–262 CrossRef CAS.
- L. F. V. Ferreira, J. P. Da Silva, I. F. Machadoa and T. J. F. Branco, Surface photochemistry: benzophenone as a probe for the study of silica and reversed-phase silica surfaces, Photochem. Photobiol. Sci., 2006, 5, 665–673 RSC.
- R. Dabestani, M. Kidder and A. C. Buchanan, Pore size effect on the dynamics of excimer formation for chemically attached pyrene on various silica surfaces, J. Phys. Chem. C, 2008, 112, 11468–11475 CrossRef CAS.
- Z. Wang, D. M. Freidrich, M. R. Beversluis, S. L. Hemmer, A. G. Joly, M. H. Heusemann, M. J. Truex, R. G. Riley and C. J. Thompson, A Fluorescence Spectroscopic Study of Phenanthrene on Porous Silica, Environ. Sci. Technol., 2001, 35, 2710–2716 CrossRef CAS.
- J. K. Thomas and E. H. Ellison, Various aspects of the constraints imposed on the photochemistry of systems in porous silica, Adv. Colloid Interface Sci., 2001, 89–90, 195–238 CrossRef CAS.
- S. A. Ruetten and J. K. Thomas, Fluorescence and triplet quantum yields of arenes on surfaces, J. Phys. Chem. B, 1998, 102, 598–606 CrossRef CAS.
- G. H. Zhang, J. K. Thomas, A. Eremenko, T. Kikteva and F. Wilkinson, Photoinduced charge-transfer reaction between pyrene and N,N′- dimethylaniline on silica gel surfaces, J. Phys. Chem. B, 1997, 101, 8569–8577 CrossRef CAS.
- D. Oelkrug, S. Reich, F. Wilkinson and P. A. Leicester, Photoionization on Insulator Surfaces - Diffuse Reflectance Laser Flash-Photolysis of Distyrylbenzenes Adsorbed on Silica and Alumina, J. Phys. Chem., 1991, 95, 269–274 CrossRef CAS.
- D. Oelkrug, S. Uhl, F. Wilkinson and C. J. Willsher, Bonding and mobility of acridine on some metal-oxide surfaces as studied by delayed fluorescence and transient absorption-spectroscopy, J. Phys. Chem., 1989, 93, 4551–4556 CrossRef CAS.
- D. Oelkrug, M. Gregor and S. Reich, Translational Mobility in the Adsorbed State - Bimolecular Deactivation Kinetics of Laser-Excited Azaaromatics on Microporous Silica, Photochem. Photobiol., 1991, 54, 539–546 CrossRef CAS.
- D. W. Bjarneson and N. O. Peterson, Direct Diffusion Measurements of Naphthacene on Silica as a Function of Silanol Density, J. Am. Chem. Soc., 1990, 112, 988–992 CrossRef CAS.
- R. Dabestani, J. Higgin, D. Stephenson, I. N. Ivanov and M. E. Sigman, Photophysical and photochemical processes of 2-methyl, 2-ethyl, and 2-tert-butylanthracenes on silica gel. A substituent effect study, J. Phys. Chem. B, 2000, 104, 10235–10241 CrossRef CAS.
- J. M. Stokke and D. W. Mazyck, Photocatalytic degradation of methanol using silica-titania composite pellets: Effect of pore size on mass transfer and reaction kinetics, Environ. Sci. Technol., 2008, 42, 3808–3813 CrossRef CAS.
- J. K. Thomas, Physical Aspects of Radiation-Induced Processes on SiO2, γ-Al2O3, Zeolites, and Clays, Chem. Rev., 2005, 105, 1683–1734 CrossRef CAS.
- L. F. V. Ferreira, A. L. Costa, I. F. Machado, A. M. do Rego, E. Sikorska and M. Sikorski, Surface photochemistry: alloxazine within nanochannels of Na+ and H+ ZSM-5 zeolites, Phys. Chem. Chem. Phys., 2009, 11, 5762–5772 RSC.
- M. Hureau, A. Moissette, S Marquis, C. Bremard and H. Vezin, Incorporation and electron transfer of anthracene in pores of ZSM-5 zeolites. Effect of Brønsted acid site density, Phys. Chem. Chem. Phys., 2009, 11, 6299–6307 RSC.
- S. Marquis, A. Moissette and C. Bremard, Incorporation of Anthracene into Zeolites: Confinement Effect on the Recombnation Rate of Photoinduced Radical Cation-Electron Pair, ChemPhysChem, 2006, 7, 1525–1534 CrossRef CAS.
- S. Marquis, A. Moissette, H. Vezin and C. Bremard, Long-Lived Radical Cation-Electron Pairs Generated by Anthracene Sorption in Non Brønsted Acidic Zeolites, J. Phys. Chem. B, 2005, 109, 3723–3726 CrossRef CAS.
- H. Vezin, A. Moissette and C. Bremard, Temperature dependant Interconversion of an Anthracene Radical Cation/Electron Moeity to an Electron-Hole Pair in the Pores of Al-ZSM-5 Zeolites, Angew. Chem., 2003, 115, 5745–5749 CrossRef.
- A. Moissette, S. Marquis, I. Gener and C. Bremard, Sorption of anthracene, phenanthrene and 9,10-dimethylanthracene on activated HZSM-5 zeolite. Effect of sorbate size on spontaneous ionization yield, Phys. Chem. Chem. Phys., 2002, 4, 5690–5696 RSC.
- L. J. Johnston, J. C. Scaiano, J. L. Shi, W. Siebrand and F. Zerbetto, Observation and Modeling of the Recombination Kinetics of Diphenylmethyl Radicals in the Cavities of Na-X Zeolite, J. Phys. Chem., 1991, 95, 10018–10024 CrossRef CAS.
- S. Hashimoto, N. Fukazawa, H. Fukumura and H. Masuhara, Observation and Characterization of Excimer Emission from Anthracene Included in Nax Zeolite, Chem. Phys. Lett., 1994, 219, 445–451 CrossRef CAS.
- S. Hashimoto, Zeolite Photochemistry: impact of zeolites on photochemistry and feedback from photochemistry to zeolite science, J. Photochem. Photobiol., C, 2003, 4, 19–49 CrossRef CAS.
- H. Garcia and H. D. Roth, Generation and reactions of organic radical cations in zeolites, Chem. Rev., 2002, 102, 3947–4007 CrossRef CAS.
- M. Alvaro, P. Ateinzar, A. Corma, B. Ferrer, H. Garcia and M. T. Navarro, Photochemical Generation of Electrons and Holes in Germanium-Containing ITQ-17 Zeolite, J. Phys. Chem. B, 2005, 109, 3696–3700 CrossRef CAS.
- S. Takagi, D. A. Tryk and H. Inoue, Photochemical Energy Transfer of Cationic Porphyrin Complexes on Clay Surface, J. Phys. Chem. B, 2002, 106, 5455–5460 CrossRef CAS.
- S. Takagi, T. Shimada, M. Eguchi, T. Yui, H. Yoshida, D. A. Tryk and H. Inoue, High-Density Adsorption of Cationic Porphyrins on Clay Layer Surfaces without Aggregation: The Size-Matching Effect, Langmuir, 2002, 18, 2265–2272 CrossRef CAS.
- M. Eguchi, S. Takagi, H. Tachibana and H. Inoue, The ‘size matching rule’ in di-, tri-, and tetra-cationic charged porphyrin/synthetic clay complexes: effect of the inter-charge distance and the number of charged sites, J. Phys. Chem. Solids, 2004, 65, 403–407 CrossRef CAS.
- S. Takagi, M. Eguchi, D. A. Tryk and H. Inoue, Light-Harvesting Energy Transfer and Subsequent Electron Transfer of Cationic Porphyrin Complexes on Clay Surfaces, Langmuir, 2006, 22, 1406–1408 CrossRef CAS.
- W. J. Albery, P. N. Bartlett, C. P. Wilde and J. R. Darwent, A General-Model for Dispersed Kinetics in Heterogeneous Systems, J. Am. Chem. Soc., 1985, 107(7), 1854–1858 CrossRef CAS.
- T. Brotin, J. P. Desvergne, F. Fages, R. Utermohlen, R. Bonneau and H. Bouas-Laurent, Photostationary fluorescence emission and time resolved spectroscopy of symmetrically disubstituted anthracenes on the meso and side rings: the unusual behaviour of the 1,4 derivative, Photochem. Photobiol., 1992, 55, 349–358 CrossRef CAS.
- D. Oelkrug, W. Honnen, F. Wilkinson and C. J. Willsher, Modeling of Transient Production and Decay Following Laser Excitation of Opaque Materials, J. Chem. Soc., Faraday Trans. 2, 1987, 83, 2081–2095 RSC.
- R. W. Kessler, G. Krabichler, S. Uhl, D. Oelkrug, W. P. Hagan, J. Hyslop and F. Wilkinson, Transient Decay Following Pulse Excitation of Diffuse- Scattering Samples, Opt. Acta, 1983, 30(8), 1099–1111 Search PubMed.
- H. K. A. Kan and T. P. Lin, Calculation of reflectance of a light diffuser with non-uniform absorption, J. Opt. Soc. Am., 1970, 60, 1252–1256 CrossRef.
- P. Lévy, Théorie de l'Addition des Variables Aléatroies, Gauthiers-Villars, Paris, 1937 Search PubMed.
- R. N. Mantegna, Lévy walks and enhanced diffusion in Milan stock exchange, Physica A, 1979, 179, 232–242 CrossRef.
- P. Barthelemy, J. Bertolotti and D. S. Wiersma, A Lévy flight for light, Nature, 2008, 453, 495–498 CrossRef CAS.
- T. J. F. Branco, A. M. Botelho do Rego, I. F. Machado and L. F. V. Ferreira, Luminescence Lifetime Distributions Analysis in Heterogeneous Systems by the Use of Excel's Solver, J. Phys. Chem. B, 2005, 109, 15958–15967 CrossRef CAS.
- R. Dabestani, K. J. Ellis and M. E. Sigman, Photodecomposition of anthracene on dry surfaces: products and mechanism, J. Photochem. Photobiol., A, 1995, 86, 231–239 CrossRef.
- G. Gery, H Fukumura and H. Masuhara, Laser Implantation of Anthracene Molecules into Poly(alkyl methacrylate) Films of Different Glass Transition Temperatures, J. Phys. Chem. B, 1997, 101, 3698–3705 CrossRef CAS.
- P. P. Levin, S. M. B. Costa and L. F. Vieira Ferreira, Pore Size Effect on Kinetics of Photoinduced Electron Transfer in the Quinone–Amine System on the Silica Surface Studied by Diffuse-Reflectance Laser Flash Photolysis, J. Phys. Chem., 1996, 100, 15171–15179 CrossRef CAS.
- L. T. Zhuravlev, Surface characterization of amorphous silica – a review of work from the former USSR, Colloids Surf., A, 1993, 74, 71–90 CrossRef.
- E. S. Pysh and N. C. Yang, Polarographic oxidation potentials of aromatic compounds, J. Am. Chem. Soc., 1963, 85, 2124–2130 CrossRef.
| This journal is © The Royal Society of Chemistry and Owner Societies 2011 |