A (6–4) photoproduct of 5-bromouracil†
Martin D.
Shetlar
* and
Vladimir J.
Basus‡
Department of Pharmaceutical Chemistry, School of Pharmacy, S-926, University of California, San Francisco, CA 94143-0446, USA. E-mail: shetlar@cgl.ucsf.edu
First published on 26th October 2010
Abstract
The photoreactions of 5-bromouracil (BrUra) induced by 254 nm radiation have received a significant amount of scrutiny over the years, both when induced in liquid aqueous solution and in the frozen state. The characterized photoproducts in the liquid state include uracil, 5,5′-diuracil (I) and a variety of products arising from ring opening, while the photoreactivity of BrUra in ice using 254 nm radiation is very low. During examination of the photoreactions induced in BrUra in frozen aqueous solution by exposure to predominately UVB irradiation, we found that several products are formed. One of these products has an ultraviolet absorption spectrum similar to those associated with (6–4) adducts of the pyrimidine bases (λmax = 326 nm). Studies with 1H and 13C nuclear magnetic resonance spectroscopy and with mass spectrometry yielded data consistent with this compound indeed being a (6–4) product, namely 6-4′-(5′-bromopyrimidin-2′-one)-5,5-dihydroxy-5,6-dihydrouracil (IIa). The product IIa probably arises from an unstable precursor, namely 5-bromo-6-4′-(5′-bromopyrimidin-2′-one)-5-hydroxy-5,6-dihydrouracil (IV); this compound is a bromohydrin and, as such, is likely unstable to debromination to 6-4′-(5′-bromopyrimidin-2′-one)-5-oxo-5,6-dihydrouracil (V). Rehydration at the 5-position to form a gemdiol would lead to IIa, the isolated product.
Introduction
The photochemistry of nucleic acids and their components is a topic of significant interest to those studying the deleterious effects of ultraviolet light on living systems. Indeed, a significant body of literature exists concerning the photochemical reactions of the nucleobase, nucleoside and nucleotide components of DNA and RNA, as well as on the photoreactions of the nucleic acids themselves. (For a comprehensive review, see Cadet and Vigny;1 updates with references to more recent publications are given in Ravanat et al.,2 while reviews by Cadet et al.3,4 discuss photoreactions induced within cellular DNA.)The photochemistry of nucleic acids containing unnatural bases (e.g. halogenated uracils) has also received study. For example, the photoreactions of 5-bromouracil (BrUra) have interested both photochemists and photobiologists for over 40 years. Photochemical studies have been conducted at the nucleobase and nucleoside level, as well as at the oligonucleotide and nucleic acid level, and a number of photoreactions have been cataloged (for reviews, see Cadet and Vigny,1 Saito and Sugiyama5 and Wang6). One of the main reasons for the interest of photobiologists in this photochemistry is the observation that incorporation of BrUra into DNA sensitizes the nucleic acid backbone to formation of products that can act as precursors to strand cleavage and cross-linking; accompanying this backbone modification is conversion of BrUra to uracil (Ura) or uracil-containing products. This aspect of BrUra photochemistry has proven of value in the study of DNA damage processes induced by irradiation of cells containing DNA in which BrUra has been partially substituted for thymine (Thy). The initial stage of such damage often involves production of the uracil-5-yl radical. Subsequent reactions within a DNA context, including hydrogen abstraction and/or oxidative processes, leads to various types of observable products in the photoreacted DNA. Strand breaks within the backbone of DNA are a common result of formation of uracil-5-yl at sites of BrU incorporation (see, for example, Chen et al.7 and Cook et al.8). Cross-linking within and between DNA strands has also been observed. For example, Cecchini et al.9 have found that interstrand cross-links can be formed in regions of BrUra-substituted DNA that contain a “bulge”, while Zeng and Wang10 have studied the nature and sequence dependence of the intrastrand cross-links formed in BrUra-substituted DNA. The photoinduced dehalogenation of 5-bromouracil (and 5-iodouracil) has been utilized to probe structural aspects of a variety of DNA structures (for a review, see Xu and Sugiyama11). For general reviews of early work on the photochemistry of DNA containing halogenated uracil, see Hutchinson;12,13 for a comprehensive recent overview of a variety of recent results on the photochemistry of 5-halouracil substituted DNA and oligonucleotides, see Tashiro and Sugiyama.14
A second reason that photobiologists have been interested in the study of the photochemistry of BrUra-substituted DNA is that irradiation of complexes of various proteins with such DNA often leads to cross-linking of the protein partner to DNA (for reviews of early work, see Saito and Sugiyama5 and Shetlar15). The potential for such cross-linking as a structural tool for probing nucleobase-amino acid contacts in nucleic acid-protein complexes has been explored in a number of studies (for a review, see Meisenheimer and Koch16).
Photochemical reactions of BrUra17,18 and 1,3-dimethylbromouracil (Me2BrUra)19 in dilute aqueous solution, using 254 nm light, produce a variety of products; in the case of BrUra the main products include such compounds as uracil, 5,5′-diuracil (I in Scheme 1), oxalic acid, urea, parabanic acid and glyoxal. In general, these reactions proceed at a slow rate. However, when BrUra is irradiated in solutions containing good hydrogen donors, then photoreaction occurs much more rapidly. A number of studies have shown that such irradiations result in formation of uracil as the principal nucleobase product;20,21 leading references to more recent work are given in Görner.22 The mechanism of this reaction appears to involve both an ion-radical pathway (via the triplet state of BrUra) and a free radical path (via the singlet state).23 Studies of the acetone sensitized reaction of BrUra in aqueous 2-propanol indicate that a chain mechanism is involved in the photoreduction reaction in this situtation.22
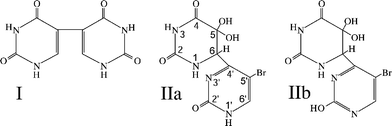 | ||
| Scheme 1 Structures of 5,5′-diuracil (I), 6-4′-(5′-bromopyrimidin-2′-one)-5,5-dihydroxy-5,6-dihydrouracil (IIa) and its lactim tautomer (IIb). | ||
Studies of the photoreactions of 5-bromouridine (BrUrd) and 5-bromo-2′-deoxyuridine (BrdUrd) have also been conducted in the frozen aqueous state, using 254 nm light to induce reaction. The main products were to shown to be 5,5′-diuridyl and 5,5′-di-2′-deoxyuridyl repectively.24 Earlier, Smith25 found that irradiation of the parent nucleobase BrUra, labeled with 82Br, in the frozen state with 254 nm light, did not lead to products containing radioactivity. Sasson and Wang24 indicated that irradiation of BrUra in the frozen state, under conditions similar to those used to study BrUrd and BrdUrd, led to very little or no decomposition of the parent compound.
One question that can be asked is whether photoproducts formed on the nucleobase or nucleoside level (other than uracil) are also produced when oligonucleotides and polynucleotides containing BrUra are irradiated. Indeed, it has been found that this is the case for one of these products. Extending earlier work,26–28 it was found that 5,5′-diuridyl was the major product resulting from irradiation of poly-rBrU.29 More recently, Gao and Wang have demonstrated that a similar type of 5-5′ cross-link involving cytosines was formed when 5-BrdCpdC, dCp5-BrdCpdA, and dCp5-BrdCdT were irradiated in solution.30
When BrUra and its nucleosides are irradiated in the presence of amino acids or related compounds, a variety of interesting reactions occur. Among these are formation of conjugates in which linkage to the amino acid replaces the Br atom at the 5-position of the uracil moiety (for a review, see Meisenheimer and Koch16). The most extensively studied systems are those in which photoinduced reaction of BrUra or related compounds with tryptophan or its derivatives occurs. For example, Saito et al.31,32 found that photoinduced coupling of BrUra and related compounds with tryptophan or its derivatives could be induced, both in the liquid phase and in the frozen state. Similarly, Dietz and Koch33 found that laser irradiation of aqueous liquid solutions containing BrUra and derivatives of tryptophan, tyrosine or histidine led to coupling between these compounds. Following up on earlier work by Varghese,34 Dietz and Koch35 confirmed that BrUra could be coupled to cysteine and, in addition, that this reaction was mediated by intermediate formation of cystine. Further work using a peptide bond model indicated that photocross-linking of BrUra to the peptide linkage could be induced.36 Reactions in which bromine is retained at the 5-position have also been studied. Photoreaction of BrUra and related compounds (e.g.5-bromo-2′-deoxyuridine) with alkyl amines leads to ring opened products. For example, irradiation of BrUra in the presence of ethylamine at basic pH yields the Z and E isomers of N-(N′-ethylcarbamoyl)-3-amino-2-bromoacrylamide as products.37
As mentioned above, there has been interest in the past few years in using BrUra, contained in a nucleic acid or oligonucleotide, as an agent for forming photocross-links to amino acids in a protein partner; from the resultant cross-linked complexes structural information could be gathered about nucleobase-amino acid contacts, particularly those involving tyrosine, tryptophan, histidine and sulfur containing amino acids. The compounds 5-iodouracil (IUra) and 5-iodocytosine (ICyt) have also been found to be useful in such experiments. (For a comprehensive review of the use of these compounds for photocross-linking, as well as a discussion of the use of BrUra for this purpose, see Meissenheimer and Koch;16experimental methods for applying cross-linking as a structural tool have been provided by the same authors38). It should be noted that IUra and ICyt have many similarities to BrUra in their photochemistry. For example, both of these compounds readily undergo photodehalogenation, both at the nucleobase and nucleoside level13,39,40 and at the level of DNA. When contained in DNA, deiodination is accompanied by chain breakage and uracil formation; in addition, oxygen-mediated reactions can result in more extensive modification of the pyrimidine moiety in DNA.41–44
In the present paper, we describe the results from experiments designed to answer the question of whether irradiation of BrUra in frozen aqueous solution with predominately UVB radiation (rather than 254 nm radiation) could lead to products similar to those observed when other non-halogenated nucleobases are irradiated. We have found that BrUra, when irradiated under these conditions, does indeed react to form a (6–4) type conjugate, namely 6-4′-(5′-bromopyrimidin-2′-one)-5,5-dihydroxy-5,6-dihydrouracil (IIa). Formation of this general type of product has been observed in a variety of other UV-irradiated nucleobase and nucleoside systems after photolysis in the aqueous frozen state or in dry films, as well as in dinucleotides, oligonucleotides and DNA that have absorbed UV light (for reviews, see Cadet and Vigny,1 Ravanat et al.2 and Wang6). However, this type of product has not been observed previously for reactant molecules containing BrUra as a moiety. Below, we describe the isolation and characterization of this new photoproduct.
Materials and methods
General aspects
Chemicals were obtained from Sigma (St. Louis, MO) and Aldrich (Milwaukee, WI). Solvents for HPLC were from Mallinkrodt (St. Louis, MO) and Fisher (Fairlawn, NJ) while NMR solvents were from Aldrich. Preparative HPLC were done on a Shiseido Capcell 10 × 250 mm UG120 column (5 μm particle size) (Yokohama, Japan), hereafter termed column A. The column used for analytical HPLC was a Shiseido Capcell UG-120 reverse phase column (4.6 × 150 mm, 5 μm particle size), termed column B. The HPLC system used was a Rainin binary gradient pumping system (Emeryville, CA) coupled to a Hewlett–Packard 1040A diode array detector (Palo Alto, CA). Ultraviolet spectra were either measured “on the fly” using the rapid detection and data analysis capabilities of a Hewlett–Packard 1040A diode array HPLC detector or on a Hewlett–Packard HP8452A spectrophotometer (Mountain View, CA). Low resolution mass spectra were run on a Kratos MS-50 (Ramsey, NJ), while high resolution mass spectra were run on a Thermo Electron Corp. LTQ Orbitrap XL (Waltham, MA). Proton NMR spectra were obtained at 300 MHz on a General Electric QE-300 NMR spectrometer (Fremont, CA), while 13C NMR spectra were run on a 400 MHz Inova NMR spectrometer (Varian, Palo Alto, CA).Preparation and isolation of the putative (6–4) product
Irradiations were done on 500 mL portions of an aerated aqueous solution containing 1 mM BrUra; the irradiations were done in a Teflon coated baking pan (29.6 cm × 22.0 cm × 5 cm). Prior to photolysis, the solution was frozen by placing the pan on a pair of 25 cm × 25 cm dry ice blocks that had been matched in thickness and placed side by side on a styrofoam sheet (to avoid damage to the countertop); to minimize frosting during the freezing process, the pan was covered with a 15 inch × 10 inch Pyrex baking dish. In a typical irradiation, the frozen solution was irradiated for 48 min using four unfiltered UVB lamps (Spectronics BLE-1T158 from Spectroline, Westbury, NY). The lamps were mounted in pairs in Spectroline XX-15A housings and the lamp housings were balanced directly on top of the pans containing the material to be irradiated. The distance of the lowest part of each cylindrical lamp surface from the surface of the frozen solution was 5 cm. The frozen solution remained on dry ice for the duration of the irradiation. Immediately after irradiation, the solution was thawed by flotation in a pan containing hot water. Analytical reverse phase HPLC of a 500 μL sample of the resultant solution was then run. Column B (see above) was employed for this analysis, using 100% water as eluent at a flow rate of 3 mL min−1. After a typical run, comparison of peak areas from HPLC of the parent and irradiated solutions showed that about 15–18% of the parent BrUra (elution time: 3.2 min) had been consumed. (The range of values for the calculated conversion is to be expected in situations where the reflective properties of the surface of the frozen solution being irradiated may not identical for each run.) After standing overnight at 4 °C, a similar HPLC analysis indicated that the calculated percentage conversion drops significantly suggesting that unstable products are formed, as well as the putative (6–4) adduct.In a typical experiment, 950 mL of irradiated solution (out of a total of 1000 mL with 15% of parent BrUra consumed) was rotatory evaporated at 40 °C to 18 mL (including rinsing solution from the evaporation flask), chilled on ice for 15 min, filtered through a Whatman HAWP 0.45 μm filter and chromatographed isocratically in 2 mL portions on HPLC column A (see above), using a mixture of 88% water and 12% methanol flowing at 6 mL min−1. The material (termed F2) corresponding to the peak containing the putative (6–4) product, eluting between 2.6 min and 3.1 min, was collected and concentrated; BrUra eluted as a peak centered at about 5.1 min under these conditions. A small peak, corresponding to 5,5′-diuracil (I), eluted between the F2 peak and the BrUra peak. The collected F2 was rotatory evaporated to 3 mL and the UV spectrum of this sample (after 300 fold dilution) was measured. Using an approximate ε value (4940), corresponding to that determined for the corresponding thymine (6–4) adduct,45 it was estimated that about 5.2 μmol of the desired adduct was collected; this corresponds to a 7.3% yield, based on the amount of BrUra consumed as measured immediately after irradiation. (To obtain sufficient IIa for 13C NMR, this procedure was repeated until enough of the desired material had been produced.) The collected F2 was concentrated and repurified on the same column, using distilled water flowing at 6 ml min−1 as eluent. Peaks eluting between 2.7 min and 3.1 min (F2-2) and 3.1 min and 3.5 min (F2-3) were collected. The fraction F2-2 was concentrated and purified once again using the same conditions. The main impurity in F2, collected as F2-3, was identified as 5-hydroxyuracil (OHUra), based on the identity of its HPLC retention time, UV spectrum, ESI mass spectrum and 1H NMR spectrum with those of an authentic sample (obtained from Sigma).
Production and isolation of photoproduct III
The (6–4) adduct of thymine45 (III) was produced as above, with the following modifications. The concentration of the irradiated solution was 2 mM. Irradiations were done using the same lamp holders, but with Spectronics BLE-1T155 lamps (principal output at 254 nm) replacing the lamps used for the BrUra irradiations. The irradiation times were 1 min; after this time, about 14.5% of the parent T had been consumed. The irradiation time chosen was that found experimentally to produce the maximum concentration of III; it can be noted that the absolute amount of III present in the thawed photolyzed solutions decreases after this irradiation time, presumably because the precursor of III reacts photochemically to form thymine trimer in what amounts to a series reaction.46A total of 10 L of solution was irradiated; the photolyzed solution was subjected to rotatory evaporation until its volume was 52 mL. After setting on ice for 30 min, this solution was filtered and then processed in 2 mL batches on the Capcell UG120 semipreparative column as above, using an eluent of 94% water/6% methanol at 5 mL min−1. The fraction eluting between 5.2 and 6 min was collected and its volume was reduced to 3.5 mL by rotatory evaporation. Under these conditions putative “trimer” (actually a trimer hydrate6) eluted at 4.7 min, the Thy c,scyclobutane dimer at 6.5 min and Thy at around 7.5 min. The 3.5 mL of solution was chilled on ice, filtered and then injected in 500 μL batches on the same column, using 92% water/8% methanol flowing at 5 mL min−1 as eluent. The fraction eluting between 4.6 and 5.4 min was collected and was found to be pure III, as evidenced by analytical HPLC and UV spectral measurements. The amount of III isolated after this purification procedure was determined to be about 49 μmol using UV spectroscopy, as described above for the putative BrUra (6–4) product; this corresponds to a yield of 3.4%, based on consumed thymine. This purified III was used to obtain the 13C NMR spectral data, given in Table 1.
| Carbon | IIa a | III b |
|---|---|---|
| a Chemical shifts are given in ppm and are relative to TSP; JCH values are in Hz. For comparison purposes, the corresponding chemical shift data for III is also provided. b The spectrum for III was run in D2O containing an internal dioxane standard; the shifts, measured relative to the dioxane standard, were then converted to a TMS standard using the relation δTMS = δdiox − 67.4 ppm. | ||
| C2 | 151.6 (s) | 153.1 (s) or 155.9 (s) |
| C4 | 167.8 (s) or 168.4 (s) | 173.2 (s) or 173.5 (s) |
| C5 | 76.7 (s) | 68.7 |
| C6 | 71.3 (d, J = 166) | 54.7 (d, J = 147) |
| C2′ | 153.6 (s) | 153.1 (s) or 155.9 (s) |
| C4′ | 167.8 (s) or 168.4 (s) | 173.2 (s) or 173.5 (s) |
| C5′ | 93.7 (s) | 113.1 (s) |
| C6′ | 149.1 (d, J = 189) | 143.7 (d, J = 180) |
| CH3 | 10.7 (q, J = 129) | |
| CH3′ | 22.6 (q, J = 130) |
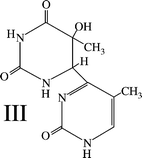 | ||
| Scheme 2 Structure of 6-4′-(5′-methylpyrimidin-2′-one)-5-hydroxy-5-methyl-5,6-dihydrouracil, the Thy(6–4)Thy adduct. | ||
It can be noted that the percentage yields of IIa (see above) and III are both single digit numbers. The predominant product produced when thymine is irradiated in ice under these conditions is the c,scyclobutane dimer; much of this compound precipitates during the rotatory evaporation process. The possible nature of the corresponding predominant product in the BrUra system will be discussed below.
Results and discussion
Identification of the putative (6–4) product contained in F2-2 as 6-4′-(5′-bromopyrimidin-2′-one)-5,5-dihydroxy-5,6-dihydrouracil (IIa)
We established the structure of the putative BrU (6–4) product to be that displayed in Scheme 1 as IIa by use of UV spectroscopy, mass spectrometry and 1H and 13C NMR spectroscopy. The UV spectrum (Fig. 1) in doubly distilled H2O shows a maximum lying between 264 nm and 360 nm that is very similar in profile to that for the corresponding (6–4) product of thymine and that is characteristic of the absorption spectrum of pyrimidin-2-ones.45 The value of this λmax is 326 nm; the spectrum also shows a second maximum at 222 nm and a minimum at 264 nm. The value of A326/A264 is 7 while the value of A222/A326 is 3.8.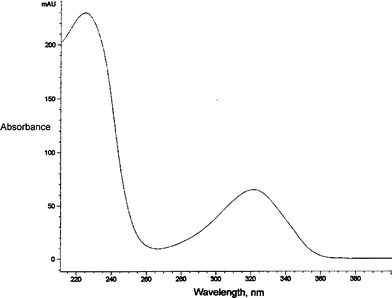 | ||
| Fig. 1 The UV spectrum of IIa, run in doubly distilled water. | ||
The electrospray ionization (ESI) mass spectrum gave a peak at 319.0, corresponding to that expected for protonated IIa; peaks were also present at 341.0 (corresponding to [IIa + Na+]) and at 356.9 [IIa + K+]. Each of these three peaks was accompanied by a partner of slightly lower intensity and two units higher in mass; this corresponds to the behavior expected for a compound containing one bromine atom and eliminates structures containing two bromine atoms. The high resolution ESI mass spectra corresponding to the two partner peaks were also consistent with protonated IIa having molecular formula of C8H8N4O5Br (peak 1: calculated: 318.9673; observed: 318.9683 and peak 2: calculated: 320.9652; observed: 320.9663).
The 1NMR and 13C NMR spectra are also consistent with this structural assignment as IIa. The proton NMR shows the following peaks in dry d6-DMSO with added TMS: δ = 5.32 (incompletely resolved quartet, C6H, J = 4.8 Hz, J′ = 3 Hz), 6.48(s,1, O5H), 6.56 (d,1, O5H, J = 4.8 Hz); 8.04(d,1,N1H, J = 3 Hz); 8.36 (broad, 1H, C6′H), 10.37 (s,1,N3H); 12.35(very broad,1,N1′H). In more moist d6-DMSO, the peak at 6.48 became significantly broader than the corresponding peak in the dry DMSO.
The peaks obtained by running the undecoupled C13 NMR spectrum of IIa in D2O, along with their assignments, are given in Table 1. In support of these assignments, the following comments can be made. The large 1JCH coupling constants associated with the peaks at δ71.3 and 149.1 indicate that the corresponding carbons must be covalently bound to hydrogen. In general, saturated carbons have smaller chemical shifts than those involved in vinyl linkages. Therefore, the 71.3 peak can be assigned as corresponding to C6 and the 149.1 as C6′. The 76.7 peak and 93.7 peaks are both singlets and therefore must correspond to a carbon without attached hydrogen. However, the bromo group is attached to a vinyl linkage, which favors the 93.7 resonance being assigned to that corresponding to C5′ and, by elimination, 76.7 as corresponding to the resonance of the C5 carbon. It can be noted that the C5 resonance in BrUra (to be compared to C5′ in IIa) occurs at δ94.4447 (see Spectrum No. 3775CDS-01-702). The remaining peaks must correspond to carbons participating in either carbonyl or imine functionalities. In general, carbonyls located at the 4-position of either saturated or unsaturated uracils have larger δ values than resonances for C2. (In dihydrouracil, C2 has a resonance at 153.9047 (see Spectrum No. 15319CDS-13-550), while in BrUra, the corresponding C2 resonance is at 150.79.) This would suggest that the resonances at 151.6 and 153.6 correspond to C2 and C2′. This implies that the remaining two resonances at δ167.8 and 168.4 correspond to the C4 and C4′ resonances; definite assignments of these last two pairs of resonances cannot be made.
Evidence that the above assignments are reasonable can be obtained by examination of the corresponding 13C NMR spectrum of the Thy(6–4)Thy adduct III. The chemical shift and spin–spin splitting data for this compound is also given in Table 1. (This set of data for III is an expanded version of that given by Shetlar and Basus48 (Table S-1 in Supplementary Data)). As can be seen, the chemical shift values for the corresponding carbons in IIa and III are reasonably close in value, except for C5′, which has a bromine attached in IIa (δ = 93.7), but a methyl group in III (δ = 113.1). In thymine, C5 has a chemical shift of 110.947 (see Spectrum No. 1089CDS-03-108).
Suggestive evidence that the pyrimidone ring in IIa may exist as a mixture of lactam and lactim forms
As mentioned above, when the proton NMR spectrum was run in very dry d6-DMSO both of the 5-hydroxyl protons gave very sharp peaks. When the spectrum was run in less carefully dried d6-DMSO, the peak at 6.48 was broad and that at 6.56 remained sharp. This suggests that the proton corresponding to the former peak is available for hydrogen bonding with water in the moister system, while the latter peak corresponds to a proton involved in internal hydrogen bonding. Another interesting observation was made in the same spectrum, namely that the proton peak corresponding to C6′ (δ8.37) was abnormally broad, while that assigned to N1′ (δ12.35) was very broad. These observations can be understood if it is postulated that (a) one of the 5-OH hydrogens is involved in internal hydrogen bonding to an oxygen or nitrogen within the molecule and (b) that the N1′ and C6′ hydrogens are found in an environment that changes over the time scale of the transitions of these protons from one spin state to another (e.g. via a tautomerization reaction converting the pyrimidone ring from a lactam form to a lactim form, in which the proton at N1′ of IIa is transferred to O2′ to form IIb).To gain some insight as to whether such ideas could have validity, we used MacSpartan Pro software (Wavefunction, Inc., Irvine, CA) to calculate the ground state energies and predict the equilibrium geometries of IIa and its tautomeric form IIb. The results of these calculations support this interpretation, indicating that about 57% of the (6–4) product would be lactam and the rest would be in the form of lactim. The results are discussed in more detail in Supplement 1, contained in the ESI.† Also included in the ESI (Fig. S-1) are ball and stick and space filling structural representations of the equilibrium geometries of IIa and IIb that were obtained via these calculations. The methodology of the calculations is discussed in the caption to Fig. S-1.
A plausible mechanism for formation of IIa
What can be said about the mechanism of formation of IIa? In general, it is thought that (6–4) adducts in nucleobase and related systems are formed via thermal rearrangement of oxetanes or azetidines, which are the primary photoproducts (see, for example, Wang6 and Cadet and Vigny1 for discussions). Photoinduced oxetane formation likely occurs within the environment provided by microcrystallites of the nucleobase or related compounds; these are formed when the nucleobase is forced out of solution by the freezing of water to form a pure solid phase (see Montenay-Garestier et al.49 for a discussion of photoreactions occurring in frozen aqueous solution).It is very likely that IIa is not the initial product produced by oxetane rearrangement, but instead results from thermal reaction of this initial rearrangement product. We suggest the mechanism given in Scheme 3 as a plausible pathway leading from a (6–4) adduct containing two bromines (IV), putatively formed by rearrangement of a parent oxetane, to the observed final product IIa. The compound IV is a bromohydrin; members of this class of compounds are known to be often unstable. Decomposition of IV to an intermediate dicarbonyl compound (V), followed by hydration would lead to IIa. Indeed, it is interesting to note that other dicarbonyl compounds are susceptible to hydration. For example, a significant fraction of biacetyl in aqueous solution is present in the hydrated form.50,51
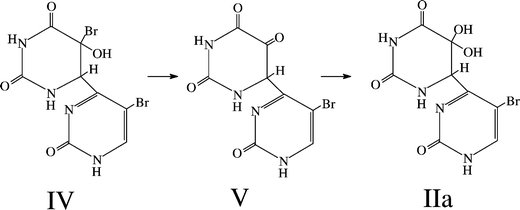 | ||
| Scheme 3 A possible sequence of reactions leading from an initially formed BrU(6–4)BrU adduct to IIa, the isolated product. | ||
Evidence for another unstable primary product
The yield of isolated IIa (about 7%), based on consumed BrUra (15%, measured immediately after thawing the irradiated BrUra solution) is not large. Obviously, other photoreactions must dominate the photochemistry in this system. (Only very minor amounts of I were produced in our experiments, indicating that reaction to produce this compound cannot account for the rest of the BrUra consumed.) However, another photoreaction of major importance has been observed. If the magnitude of the loss of BrUra is measured after the thawed solution is allowed to stand at room temperature overnight, it is found to be considerably smaller than if measured directly after irradiation. Relevant to this observation of decreased degree of loss of BrUra upon standing is yet another observation. Using the analytical chromatography conditions described under Materials and methods, a large peak elutes after BrUra in the HPLC chromatogram of freshly thawed irradiated BrUra solution; indeed this peak tails into the BrUra peak under these conditions, suggesting that the compound is decomposing on the column during chromatography. Consistent with this interpretation, this product peak was greatly diminished in size for irradiated BrUra solutions that had thawed and stood overnight at room temperature or, alternatively, that had been subjected to rotatory evaporation at temperatures elevated above room temperature. Furthermore, the compound responsible for the corresponding HPLC peak disappeared completely from irradiated solutions that were kept in the cold at all times if the time of standing was sufficiently long. In all cases, the peak area for the BrUra peak in such solutions was observed to increase significantly. Likewise, rotatory evaporation to dryness at room temperature greatly reduced the amount of this compound with concomitant production of BrUra. While we have been successful in isolating this product in a dilute state, its stability properties have made it very difficult to keep it in a pure state under those conditions required for sample preparation for NMR and mass spectrometry. The one piece of solid spectral information we do have is the UV spectrum; however, it is not terribly informative concerning the detailed structure. The spectrum shows absorbance between 210 and 300 nm, but does not display a λmax; this suggests that all carbon–carbon bonds in the structure of the compound are saturated. One possibility is that the unstable material is cyclobutane dimer in nature, perhaps trans-anti (t,a) in structure. It is known that the t,acyclobutane dimer of Thy is particularly unstable with respect to reversion to Thy upon heating. The half-life for reversion of the t,a dimer of thymine to thymine at 100 °C in neutral solution is just a few minutes (see, for example, Fig. 11 in Fisher and Johns52). Further experimentation will be required to determine more about the nature of this unstable major photoproduct.Secondary reactions of IIa: exploratory studies
We have conducted some exploratory investigations of the photochemical and thermal reactivity of IIa; the results are discussed below.As expected, increasing the temperature of incubation above 37 °C increased the rate of disappearance of IIa and lowering it decreased the rate of decomposition. After 17 h incubation of IIa at 27 °C the percentage of starting material consumed was 6%, while after 16 h at 42 °C the corresponding percentage was 46%. Only 3 h was required to achieve 50% loss of starting material at 55 °C.
The decomposition of IIa to form products having absorption spectra similar to that of the parent is mimicked when the Thy(6–4)Thy adduct (III) is similarly treated. When a sample of III was incubated at 55 °C for 20 h and analyzed using the same HPLC conditions as employed above, it was found that the peak area (measured at 220 nm) corresponding to III decreased by 16%; the elution time for III was 2.4 min under these conditions. Six new peaks with absorption spectra similar to III were evident in the HPLC chromatogram. These compounds eluted at 1.4 min, 1.7 min, 2.0 min, 3.1 min, 5.2 min and 6.6 min with the 3.1 min peak being dominant, the 1.4 min peak being second in importance and the rest having smaller peak areas; a small peak eluting at 5.7 min had an absorption spectrum similar to that of the dehydration product of III (see Figure 4 in Wang6). The thermal instability of (6–4) adducts has been noted before. In a study of the (6–4) adducts of thymidine, Shaw56 (in his Ph. D. thesis, done in the laboratory of Jean Cadet in Grenoble) noted that these compounds degrade thermally upon standing to produce secondary products with spectra similar to the primary photoproducts.
Summarizing, the results of the exploratory experiments described above suggest that IIa (and III) are thermally labile in aqueous solution towards production of degradation products. In the case of IIa, these products include parent BrUra and OHUra. For both IIa and III, a number of products are formed that have UV absorption spectra that are characteristic of (6–4) adducts (i.e. a strong maximum in absorbance above 300 nm). The presence of this absorbance maximum, characteristic of the pyrimidone moiety in these species, indicates that these decomposition products probably retain this moiety and that the degradative reactions have occurred in the dihydropyrimidine portion of the molecule. Irradiation of IIa with UVB results in formation of new products without appreciable absorbance above 250 nm; however, upon irradiation of photoreaction mixtures containing these compounds with 254 nm light, they decompose to new products that don't have appreciable absorbance above 250 nm, rather than undergo photoreversal to the original IIa. Further work will be needed to completely characterize the products and elucidate the pathways of formation of the various types of reactions that occur when IIa is incubated at room temperature and above or when it is irradiated with UVB light.
Concluding remarks
The results of previous work on the photolysis of BrUra in the frozen state using 254 nm radiation indicated that bromine containing products were not formed25 and observed that BrUra was very resistant to chemical change under these conditions.24 Above, we have described the isolation of the (6–4) product IIa that is produced as a result of irradiation of BrUra in frozen solution at dry ice temperature. This is the first reported example of a (6–4) adduct being produced via irradiation of a halogenated nucleobase. Key to observation of this compound was the use of a light source with output predominately in the UVB region of the spectrum. At least one other major unstable product is also formed; its nature remains to be determined.Abbreviations
| BrUra | 5-Bromouracil |
| BrdUrd | 5-Bromo-2′-deoxyuridine |
| BrUrd | 5-Bromouridine |
| DMSO | Dimethylsulfoxide |
| ESI | Electrospray ionization |
| HPLC | High performance liquid chromatography |
| MeOH | Methanol |
| OHUra | 5-Hydroxyuracil |
| Thy | Thymine |
| TMS | Tetramethylsilane |
| TSP | 3-(Trimethylsilyl)propionic-2,2,3,3-d4 acid, sodium salt |
| Ura | Uracil |
Acknowledgements
Research support from the National Science Foundation (CHE-0131203) is gratefully acknowledged. We also gratefully acknowledge the Bio-organic, Biomedical Mass Spectrometry Resource (A. L. Burlingame, Director), supported by the NIH Division of Research Resources Grant RR 01614 and, especially, David Maltby who took special pains to obtain needed mass spectra in a timely manner.References
- J. Cadet, and P. Vigny, The photochemistry of nucleic acids, in Bioorganic Photochemistry, Vol. 1, ed. H. Morrison, John Wiley and Sons, New York, 1990, pp. 1–272 Search PubMed.
- J.-L. Ravanat, T. Douki and J. Cadet, Direct and indirect effects of UV radiation on DNA and its components, J. Photochem. Photobiol., B, 2001, 63, 88–102 CrossRef CAS.
- J. Cadet, S. Courdavault, J.-L. Ravanat and T. Douki, UVB and UVA radiation-mediated damage to isolated and cellular DNA, Pure Appl. Chem., 2005, 77, 947–961 CrossRef CAS.
- J. Cadet, E. Sage and T. Douki, Ultraviolet radiation-mediated damage to cellular DNA, Mutat. Res., 2005, 571, 3–17 CrossRef CAS.
- I. Saito, and H. Sugiyama, Photoreactions of nucleic acids and their constituents with amino acids and related compounds, in Bioorganic Photochemistry, Vol. 1, ed. H. Morrison, John Wiley and Sons, New York, 1990, pp. 317–340 Search PubMed.
- S. Y. Wang,Pyrimidine bimolecular photoproducts, in Photochemistry and Photobiology of Nucleic Acids, Vol. 1, ed. S. Y. Wang, Academic Press, New York, 1976, pp. 295–356 Search PubMed.
- T. Q. Chen, G. P. Cook, A. T. Koppisch and M. M. Greenberg, Investigation of the origin of the sequence selectivity for the 5-halo-2 ′-deoxyuridine sensitization of DNA to damage by UV-irradiation, J. Am. Chem. Soc., 2000, 122, 3861–3866 CrossRef CAS.
- G. P. Cook, T. Q. Chen, A. T. Koppisch and M. M. Greenberg, The effects of secondary structure and O-2 on the formation of direct strand breaks upon UV irradiation of 5-bromodeoxyuridine-containing oligonucleotides, Chem. Biol., 1999, 6, 451–459 CrossRef CAS.
- S. Cecchini, C. Masson, C. La Madeleine, M. A. Huels, L. Sanche, J. R. Wagner and D. J. Hunting, Interstrand cross-link induction by UV radiation in bromodeoxyuridine-substituted DNA: Dependence on DNA conformation, Biochemistry, 2005, 44, 16957–16966 CrossRef CAS.
- Y. Zeng and Y. Wang, Sequence-dependent formation of intrastrand crosslink products from the UVB irradiation of duplex DNA containing a 5-bromo-2 ′-deoxyuridine or 5-bromo-2 ′-deoxycytidine, Nucleic Acids Res., 2006, 34, 6521–6529 CrossRef CAS.
- Y. Xu and H. Sugiyama, Photochemical approach to probing different DNA structures, Angew. Chem., Int. Ed., 2006, 45, 1354–1362 CrossRef CAS.
- F. Hutchinson, The lesions produced by ultraviolet light in DNA containing 5-bromouracil, Q. Rev. Biophys., 1973, 6, 201–246 CAS.
- F. Hutchinson and W. Kohnlein, The photochemistry of 5-bromouracil and 5-iodouracil in DNA, Prog. Mol. Subcell. Biol., 1980, 7, 1–42 CAS.
- R. Tashiro, and H. Sugiyama, Reactivity of 5-halopyrimidines in nucleic acids, in Radical and Radical Ion Reactivity in Nucleic Acid Chemistry, ed. M. M. Greenberg, Wiley, New York, 2009, pp. 163–189 Search PubMed.
- M. D. Shetlar, Cross-linking of proteins to nucleic acids by ultraviolet light, Photochem. Photobiol. Rev., 1980, 5, 105–197 Search PubMed.
- K. Meisenheimer and T. Koch, Photocross-linking of nucleic acids to associated proteins, Crit. Rev. Biochem. Mol. Biol., 1997, 32, 101–140 CrossRef CAS.
- H. Ishihara and S. Y. Wang, Photochemistry of 5-bromouracils: isolation of 5,5′-diuracils, Nature, 1966, 210, 1222–1226 CAS.
- H. Ishihara and S. Y. Wang, Photochemistry of 5-bromouracil in aqeuous solution, Biochemistry, 1966, 5, 2307–2313 CrossRef CAS.
- H. Ishihara and S. Y. Wang, Photochemistry of 5-bromo-1,3-dimethyluracil in aqueous solution, Biochemistry, 1966, 5, 2302–2307 CrossRef CAS.
- J. M. Campbell, D. Schulte-Frohlinde and C. von Sonntag, Quantum yields in UV photolysis of 5-bromo-uracil in presence of hydrogen donors, Photochem. Photobiol., 1974, 20, 465–467 CrossRef CAS.
- J. M. Campbell, C. von Sonntag and D. Schulte-Frohlinde, Photolysis of 5-bromouracil and related compounds in solution, Z. Naturforsch. B, 1974, 29B, 750–757.
- H. Görner, Free-radical chain mechanism in the acetone-sensitized photolyis of 5-bromouracil and derivatives in alcohol-water mixtures, J. Photochem. Photobiol., A, 1995, 89, 147–156 CrossRef.
- B. J. Swanson, J. C. Kutzer and T. H. Koch, Photoreduction of 5-bromouracil. Ionic and free radical pathways, J. Am. Chem. Soc., 1981, 103, 1274–1276 CrossRef CAS.
- S. Sasson and S. Y. Wang, Photochemistry of 5-bromouridine and 5-bromo-2′-deoxyuridine in ice and in puddles, Photochem. Photobiol., 1977, 26, 357–361 CrossRef CAS.
- K. C. Smith, Photochemical reactions of thymine, uracil, uridine, cytosine and bromouracil in frozen solution and in dried films, Photochem. Photobiol., 1963, 2, 503–517 CrossRef CAS.
- M. Ehrlich and M. Riley, Photolysis of polyribobromouridylic acid, Photochem. Photobiol., 1972, 16, 385–395 CrossRef CAS.
- M. Ehrlich and M. Riley, Oligonucleotide photoproducts formed by photolysis of polyribobromouridylic acid, Photochem. Photobiol., 1972, 16, 397–412 CrossRef CAS.
- M. Ehrlich and M. Riley, Effect of base sequence on ultraviolet-irradiation products of double-stranded polynucleotides containing bromouracil and adenine, Photochem. Photobiol., 1974, 20, 159–165 CrossRef CAS.
- S. Sasson, S. Y. Wang and M. Ehrlich, 5,5′-diuridynyl, a major photoproduct from UV-irradiation of polynucleotides containing bromouracil, Photochem. Photobiol., 1977, 25, 11–13 CrossRef CAS.
- Y. Gao and Y. Wang, The photochemistry of 5-BrdCpdC, dCp5-BrdCpdA, and dCp5-BrdCdT in aqueous solution, Nucleosides, Nucleotides Nucleic Acids, 2006, 25, 279–287 CrossRef CAS.
- I. Saito, S. Ito and T. Matsuura, Photoinduced coupling reaction of 5-bromouridine to tryptophan derivatives, J. Am. Chem. Soc., 1978, 100, 2901–2902 CrossRef CAS.
- I. Saito, S. Ito, T. Matsuura and C. Hélène, Specific photocoupling of 5-bromouridine to tryptophan in aqueous frozen solutions, Photochem. Photobiol., 1981, 33, 15–19 CrossRef CAS.
- T. M. Dietz and T. H. Koch, Photochemical coupling of 5-bromouracil to tryptophan, tyrosine and histidine, peptide-like derivatives in aqueous fluid solution, Photochem. Photobiol., 1987, 46, 971–978 CAS.
- A. J. Varghese, Photoreactions of 5-bromouracil in the presence of cysteine and glutathione, Photochem. Photobiol., 1974, 20, 461–464 CrossRef CAS.
- T. M. Dietz and T. H. Koch, Photochemical reduction of 5-bromouracil by cysteine derivatives and coupling of 5-bromouracil to cystine derivatives, Photochem. Photobiol., 1989, 49, 121–129 CrossRef CAS.
- T. M. Dietz, R. J. von Trebra, B. J. Swanson and T. H. Koch, Photochemical coupling of 5-bromouracil (BU) to a peptide linkage. A model for BU-DNA protein photocrosslinking, J. Am. Chem. Soc., 1987, 109, 1793–1797 CrossRef CAS.
- M. D. Shetlar, R. B. Rose, K. Hom and A. A. Shaw, Ring opening photoreactions of 5-bromouracil and 5-bromo-2′-deoxyuridine with selected alkylamines, Photochem. Photobiol., 1991, 53, 595–609 CrossRef CAS.
- K. M. Meisenheimer, P. L. Meisenheimer and T. H. Koch, Nucleoprotein photo-cross-linking using halopyrimidine-substituted RNAs, Methods Enzymol., 2000, 318, 88–104 CAS.
- H. Görner, Acetone-sensitized photolysis of 5-iodouracil and 5-iodouridine in aqueous solution in the presence of alcohols-free radical chain mechanism, J. Photochem. Photobiol., A, 1993, 72, 197–208 CrossRef.
- R. O. Rahn, Photochemistry of halogen pyrimidines-iodine release studies, Photochem. Photobiol., 1992, 56, 9–15 CrossRef CAS.
- R. O. Rahn and R. S. Stafford, Photochemistry of DNA containing iodinated cytosine, Photochem. Photobiol., 1979, 30, 449–454 CrossRef CAS.
- R. O. Rahn and H. G. Sellin, Photolysis of iododeoxycytidine in DNA and related polynucleotides-wavelength dependence and energy transfer, Photochem. Photobiol., 1980, 32, 313–317 CrossRef CAS.
- R. O. Rahn and H. G. Sellin, Action spectra for the photolysis of 5-iododeoxyuridine in DNA and related model systems-evidence for short-range energy transfer, Photochem. Photobiol., 1982, 35, 459–465 CrossRef CAS.
- R. O. Rahn and H. G. Sellin, Chain breakage accompanying the photolysis of IdUrd-DNA, Photochem. Photobiol., 1983, 37, 661–664 CAS.
- A. J. Varghese and S. Y. Wang, Thymine-thymine adduct as a photoproduct of thymine, Science, 1968, 160, 186–187 CrossRef CAS.
- R. O. Rahn and J. L. Hosszu, Photochemical studies of thymine in ice, Photochem. Photobiol., 1969, 10, 131–137 CrossRef CAS.
- National Institute of Advanced Industrial Science and Technology (AIST), Spectral Database for Organic Compounds (SDBS), access at: http://riodb01.ibase.aist.go.jp/sdbs/cgi-bin/cre_index.cgi?lang=eng.
- M. D. Shetlar and V. J. Basus, The photochemistry of uracil: A reinvestigation, Photochem. Photobiol. DOI:10.1111/j.1751-1097.2010.00826.x.
- T. Montenay-Garestier, M. Charlier, and C. Hélène, Aggregate formation, excited-state interactions and photochemical reactions in frozen aqueous solutions of nucleic acid constituents, in Photochemistry and Photobiology of Nucleic Acids, Vol. 1, ed. S. Y. Wang, Academic Press, New York, 1976, pp. 381–417.
- Y. Lee and J. G. Burr, Emission from the hemihydrate species CH3COC(OH)2CH 3 in aqueous solutions of biacetyl, Chem. Phys. Lett., 1976, 43, 146–148 CrossRef CAS.
- K. Miyata, K. Nakashima and M. Koyanagi, Spectroscopic evidence of α,α-hydroxyketone in biacetyl aqeuous solution, Bull. Chem. Soc. Jpn., 1989, 62, 367–371 CrossRef CAS.
- G. J. Fisher, and H. E. Johns, Pyrimidine photodimers, in Photochemistry and Photobiology of Nucleic Acids, Vol. 1, ed. S. Y. Wang, Academic Press, New York, 1976, pp. 225–294.
- J.-S. Taylor, D. S. Garrett and M. P. Cohrs, Solution-state structure of the Dewar pyrimidinone photoproduct of thymidylyl-(3′-5′)-thymidine, Biochemistry, 1988, 27, 7206–7215 CrossRef CAS.
- B. P. Ruzsicska, and D. G. E. Lemaire, DNA photochemistry, in CRC Handbook of Organic Photochemistry and Photobiology, ed. W. M. Horspool and P.-S. Song, CRC Press, Boca Raton, FL, 1995, pp. 1289–1315 Search PubMed.
- L. Celewicz, M. Mayer and M. D. Shetlar, The photochemistry of thymidylyl-(3′-5′)-5-methyl-2′-deoxycytidine in aqueous solution, Photochem. Photobiol., 2005, 81, 404–418 CrossRef CAS.
- A. A. Shaw, A study of the direct effects of ionising and far ultraviolet radiation on nucleic acids, Ph. D. Thesis, University of Surrey, 1987.
Footnotes |
| † Electronic supplementary information (ESI) available: Results of quantum chemical calculations for the BrUra (6–4) adduct (IIa) and its lactim tautomer (IIb). See DOI: 10.1039/c0pp00242a |
| ‡ Permanent mailing address: Caixa Postal 007, Extrema, Minas Gerais, Brazil, 37640-006. |
| This journal is © The Royal Society of Chemistry and Owner Societies 2011 |