pH and temperature dependent relaxation dynamics of Hoechst-33258: a time resolved fluorescence study
Nilotpal
Barooah
,
Jyotirmayee
Mohanty
,
Haridas
Pal
,
Sisir K.
Sarkar
,
Tulsi
Mukherjee
and
Achikanath C.
Bhasikuttan
*
Radiation & Photochemistry Division, Bhabha Atomic Research Centre, Mumbai, 400 085, India. E-mail: bkac@barc.gov.in
First published on 26th October 2010
Abstract
The photophysical behavior of Hoechst 33258 (H33258) in aqueous solution has been studied by steady-state and time-resolved fluorescence measurements. The intriguing intramolecular geometrical orientations of the dye bring out major modulation on its photophysical behavior, especially in the fluorescence emission characteristics with pH. It has been seen that a change in the solution pH from 7 to 4.5 enhances the emission yield by ∼20 fold and this change is ∼80-fold on changing the pH from 1.5 to 4.5. While a fast flipping motion among the two benzimidazole rings is considered to be one of the most probable mechanisms for the fast fluorescence decay, a more planar structure of the dicationic form at pH 4.5 having a double bond character between the two benzimidazolium groups is suggested to be the most likely fluorescent species. A similar planar structure is in fact considered to be the fluorescent emitting species of H33258 on minor groove binding to DNA. On the basis of temperature dependent fluorescence decay dynamics explored for the dye in solutions at pH 7 and 4.5, it is understood that a nearly isoenergetic double-well excited state potential is possibly involved in the excited state relaxation dynamics of the dye at pH 7. On increasing the temperature, the conversion to the planar structure is facilitated from the non-planar LE state, enhancing the emission probability of the dye.
Introduction
Luminescent probes which are responsive to perturbations due to molecular interactions or ambient conditions are well projected as molecular reporters for detection and quantification of biomolecular assays, especially for DNA and proteins.1–4 Typically, these probes are extrinsic organic dye molecules which are known to display distinctive changes in the photophysical properties on interaction and can be monitored by convenient optical techniques.4–7 Among many such molecular probes, Hoechst 33258 (H33258, Chart 1) has attracted immense research interest due to its strong groove binding interaction with DNA and is widely used in biophysical investigations of cells and quantification of DNA.8–11 H33258 belongs to the bisbenzimidazole class of molecules having anticancer properties for their ability to inhibit topoisomerase and many other cellular processes.11 H33258 and its derivatives are found to provide significant protection against radiation induced DNA strand breakage and therefore its potential as a radioprotector has been the subject of continuous investigation.12,13 It is needless to mention that all these noteworthy applications stem from the specific interaction of the dye with the DNA or polynucleotides which are observed as remarkable enhancement in its fluorescence yields.14–16 Further, the dye is known to be specific towards the minor grooves of double-stranded DNA and also for the AT-rich sequences, and has long been used for quantitative determination of double-stranded and single-stranded DNA on the basis of fluorescence intensity enhancement.14–16 Obviously, since the fluorescence parameters hold the key to the recognition and detection of the DNA/biomolecules, the understanding of the associated changes in the dye on binding to the biomolecule becomes crucial. | ||
| Chart 1 Structure of H33258. | ||
As discussed above, owing to wide ranging application of H33258, both as a probe and a drug, its photophysical properties, especially the luminescent properties, have been a subject of several investigations, both in free form and bound to DNA.17–25 It has been widely accepted that the large fluorescence enhancement on binding to the minor grooves of DNA is due to the planar structure of the dye, which is largely devoid of the non-radiative channels.22,23 It is certain that the presence of several protophilic nitrogens and their acidity constants (pKas) play a decisive role in the structural orientation and hence the emission properties of the dye. This issue has been the subject of several experimental and theoretical studies.18,20,25–27 On the other hand, it is imperative to note that the probe itself displays large fluorescence enhancement in aqueous solutions on varying the solution pH. It has been observed that ∼8-fold enhancement in the emission intensity takes place on changing the solution pH from 2 to 4.20 This is a very distinctive feature since such emission enhancement has been considered as the indicator in quantification when it interacts with DNA. It is to be noted here that different protolytic structures of the dye/drug can display large variations in the specific activity, especially when its binding/release mechanisms on the biomolecules are mainly dependent on the prototropic structures of the dye.28 It is a matter of concern that having a large number of potential proton donor groups available on biomacromolecules, a possible shift in the protolytic equilibrium cannot be ruled out completely, making a direct quantification of the fluorescence probe tricky. Though some of the earlier studies have attempted to explore this aspect, the intriguing excited state relaxation and its corresponding fluorescence properties still remain poorly explained.18,20
In this study, we revisit the absorption and emission properties of H33258 with a systematic change in the solution pH and temperature. Our observation of a significant temperature dependence on the emission at pH 7 is seen in sharp contrast to the emission data obtained for the dye solution at pH 4.5. These exciting results were explained on the basis of a double-well excited state potential with probable interconversion among different geometrical structures. These remarkable structural changes with pH and the consequent fluorescence properties are discussed in relation to its interaction with DNA.
Experimental
Hoechst 33258 (2′-4-hydroxyphenyl)-5-(4-methyl-1-1-piperazinyl)-2,5′-bi-1H-benzimidazole) tris-hydrochloride, was obtained from Sigma Chemical Co (>98% HPLC) and was used as received. Several tests were carried out at different fluorescence excitation conditions and confirmed that the dye is pure for the measurements reported here. Absorption spectra were recorded with a Shimadzu 160A spectrophotometer (Tokyo, Japan). Steady-state fluorescence spectra were recorded using a Hitachi F-4500 spectrofluorimeter (Tokyo, Japan). Nanopure water (conductivity less than 0.06 μS cm−1), obtained from a Millipore Gradiant A10 system, was used to prepare the sample solutions. Solution pH was maintained by either phosphate buffer (∼10 mM), tris buffer (∼10 mM) or HCl/KCl (∼10 mM), wherever applicable. The samples were excited at 365 nm. In all the measurements, the dye concentration was kept less than 2 μM, unless specified otherwise. The fluorescence quantum yield has been estimated by comparing the integrated corrected fluorescence spectra of H33258 with that of the standard (ϕf(Coumarin 152/acetonitrile) = 0.22)) as described elsewhere.1 Time-resolved fluorescence measurements were carried out using a time-correlated single-photon counting (TCSPC) spectrometer (IBH, UK). In the present work, a 374 nm diode laser (100 ps, 1 MHz repetition rate) was used for excitation and a MCP PMT was used for fluorescence detection. With the present setup, the instrument time resolution is adjudged to be better than 50 ps. From the measured decay traces, the time constants were evaluated following a reconvolution procedure using multi-exponential decay functions.1 Computational studies were performed with Gaussian 92 package.29Results and discussion
Steady-state absorption and fluorescence measurements
Considering the presence of several protophilic nitrogens in the structure of H33258, the absorption and emission spectral changes for the dye were monitored in aqueous solution at different preset pH conditions. Fig. 1 presents the steady-state absorption spectra of H33258 (∼2 μM) recorded from solutions maintained at pH 7, 4.5, 1.5 and 11. At pH 7, the absorption spectrum shows maximum at 338 nm and is in good agreement with that reported earlier.20 Note that the dye is known for its tendency to form aggregates at higher concentrations in aqueous solutions and the data report aggregation above 10 μM20 or 40 μM23 of dye concentration. Hence in our studies, care has been taken to maintain the dye concentration as low as possible (<2 μM) in order to avoid any erroneous results. In comparison with the profile at pH 7, the dye solution at lower and higher solution pH displayed significant hyperchromic and bathochromic shifts (∼16 nm), which could be an indication of severe changes in the electronic distribution due to different protonated structure of the dye. Though similar absorption spectral measurements have been reported earlier, the remarkable changes in the absorption coefficient with pH has not been shown convincingly, probably due to the much higher dye concentration (more than 30 μM) employed in those measurements where the dye aggregation becomes significant.20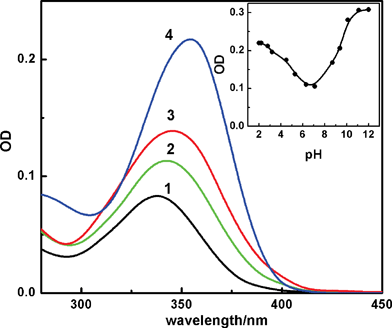 | ||
| Fig. 1 Absorption spectra of ∼2 μM of H33258 in aqueous solution at different pHs; 7 (1), 4.5 (2), 1.5 (3) and 11 (4). Inset shows the absorbance changes monitored at 360 nm with solution pH. | ||
In view of the large changes in the absorption spectra with pH, the ground state pKa was evaluated by monitoring the absorption changes at 360 nm on changing the pH and is shown in the inset of Fig. 1. The pKa curve confirms two close lying pKa values below pH 7 and can be assigned to the values 3.5 and 5.5 reported earlier.20 Recently Ladinig et al. have discussed the distribution of charged states as a function of solution pH.17 Based on combined experimental and theoretical studies, they have proposed that at pH 4.5 the dicationic species exists in its maximum probability, whereas at pH 1.5 and 7 respective tricationic and monocationic states have the major contributions. Therefore, in the pH range 1–7, the different charged states can be populated to different extent by varying the pH. So at pH 7, the monocationic species having a protonated methyl substituted piperazine nitrogen and undissociated phenolic OH is the predominant form, whereas at pH ∼1.5, it is likely that both the imidazolium nitrogens on either rings also get protonated, making majority of the dye tricationic.18,20,26 At pH 4.5, the dye exists as dicationic where the imidazolium nitrogen of the benzimidazole unit adjacent to the piperazine is protonated. All these protonation equilibria bring out marked differences in the chemical structure of the dye resulting in significant changes in the electronic charge distribution and hence the spectral features.
Similarly, in the fluorescence domain also notable changes have been observed in the emission profiles recorded on changing the solution pH and are shown in Fig. 2. As discussed earlier, at any specified pH, the fluorescence yield will be governed by the contributions from all the charged states of the dye and will evidently reflect the predominant charge states of the dye at that pH.17 At pH 7, the dye exhibited very weak fluorescence emission with λmax at ∼502 nm (trace 1, Fig. 2). The estimated quantum yield following a comparative method1 is ∼0.02, in good agreement with that reported in the literature.4,20 On decreasing the pH, the dye showed an increase in the emission intensity, reaching a maximum value at pH ∼4.5 (trace 2, Fig. 2, ϕf = 0.4). In this case, the emission profile also shifted bathochromically by ∼22 nm. However, on further acidifying the solution to pH ∼1.5, the emission intensity decreased drastically (trace 3, Fig. 2) with the emission yield showing much lower value and is about 0.005. This value is significantly lower than the value of 0.05 reported by Gorner at pH 2 and is a clear manifestation of the pH dependence on the emission intensity.20 In any case, the remarkably large fluorescence enhancement at pH ∼4 was also observed by Gorner and by several others, however, apparently the focus has been shifted towards its binding with DNA and less attention has been paid to understand the photophysics of the dye itself.20,22,23,25 We consider these changes very informative in understanding the excited state relaxation dynamics of different protonated structures of the dye and to correlate their structural orientation with their radiative/non-radiative deexcitation pathways.
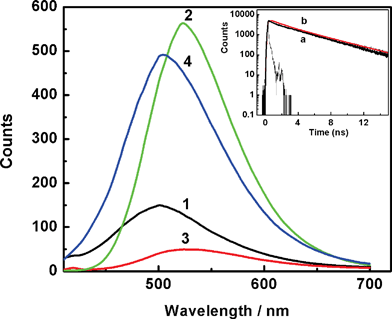 | ||
| Fig. 2 Fluorescence spectra recorded from solution at preset pH values 7 (1), 4.5 (reduced by 6-fold) (2), 1.5 (3) and 11 (4). Absorbance normalized at λexc = 365 nm. Inset shows the comparison of the fluorescence decay traces recorded for H33258 from a solution at pH 4.5 (a) and H33258 in presence of CT-DNA at pH 7 (b). | ||
As mentioned above, it is quite unusual that the emission quantum yield displays about 20-fold enhancement at pH ∼4.5 as compared to that at pH 7 and is about 80-fold enhancement in comparison with that at pH 1.5. As reported widely, large enhancement in fluorescence has been observed for H33258 on binding to AT-rich double-stranded DNA.22,23 It is believed that the dye which is a well-known minor groove binder, attains a planar structure on binding to DNA and this planarity is proposed as the main reason for the enhanced emission yield for the bound dye. However, in the present study the fluorescence measurements carried out for the free dye in aqueous solutions at different pHs also display the changes comparable with that observed in the presence of DNA. This is an intriguing observation and poses a problem in relation to quantitative measurements in the presence of DNA.18,20 Therefore, it is important to have a clear distinction between the emission behavior of different protonated structures and their characteristics. To further explore the intricacies of different protonated structures, the studies have also been extended by employing the time-resolved fluorescence measurements.
Time-resolved fluorescence measurements
Commensurate with the steady-state fluorescence measurements, the excited state dynamics of H33258 is found to be very sensitive to the solution conditions like solvent, pH, temperature etc.Fig. 3 presents the fluorescence decay traces recorded at 500 nm at different pH conditions, namely, pH 7, 4.5, 1.5 and 11. The decay profile at pH 7 (Fig. 3, trace 1) fit to a tri-exponential decay function with time constants 60 ps (15%), 365 ps (24%) and 3.8 ns (61%), and are in line with the earlier reports.22,23,25 It is realized that the fluorescence decays of the dye is inherently multi-exponential in nature, possibly representing different conformational structures of the dye in the solution which are sensitive to the solution pH. Gradual decrease in the solution pH from 7 to ∼4.5 displayed dramatic changes in the decay behavior in which the slower component increased significantly with the decay profile attaining nearly single exponential having 96% of 3.8 ns decay (Fig. 3, trace 2). However, further decrease in the solution pH to ∼1.5 again changed the decay profile displaying significant increase in the contribution of the fast component; 35 ps (47%), 534 ps (19%), 2.8 ns (34%) (Fig. 3, trace 3). On the other side at higher alkalinity, the decay trace recorded at pH 11 displayed distinctly different decay pattern (trace 4) having only the fast decay components with the time constant values 76 ps (16%), 593 ps (84%) and may correspond to a totally different relaxation channel as the phenolic OH is also expected to be deprotonated.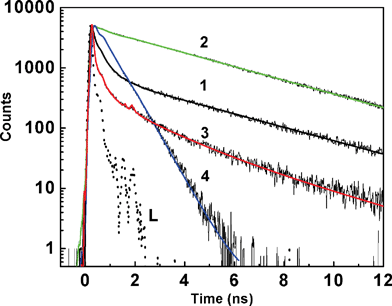 | ||
| Fig. 3 Fluorescence decay traces recorded from H33258 solution at pH; 7 (1); 4.5 (2); 1.5 (3) and 11 (4). L represents the excitation lamp profile. | ||
This is a very interesting observation as the excited state decay dynamics has been a topic of much debate to understand the unusual emission behavior observed for H33258. Among several non-radiative excited state processes like proton transferetc., an intramolecular flipping motion among the benzimidazolium rings of these protonated structure is considered as one of the prominent relaxation mechanism. Supporting evidence has been obtained when higher medium viscosity or rigid macrocyclic environments were introduced to slow down such structural movements. In the past, several attempts have been made to probe the unusual fluorescence behavior of H33258, with major focus on its dynamics when the dye is bound to the DNA, and several explanations have been proposed for the fluorescence behavior.4,21–24 The majority of the arguments were based on the basis of a torsional motion in the molecule and also on a charge transfer (CT) structure as the main reason for the de-excitation pathways. Another set of literature argues the de-excitation mechanism is due to an intramolecular proton transfer from the phenol to the benzimidazolium nitrogen, thus creating a weakly fluorescent emitting quinoid structure. The resemblance of the decay values for H33258 and its phenoxyethyl derivative at pH below 7 rules out the suggestion that non radiative deactivation of the excited dye consists of an intramolecular proton transfer from phenol to the closest benzimidazole nitrogen.4 Pal et al. have investigated the ultrafast relaxation dynamics of H33258 on DNA surface and reported ultrafast components in the hydration correlation function corresponding to the local hydration sphere.24 On a slower time scale, Pal et al. followed the excited state decay at three preset pH conditions pHs 7, 1.2 and 12, which has been extended to micelle and reverse micelle systems.25 However, no mention has been made on the fluorescence behavior at pH 4.5.25 Cosa et al. have reported excited state lifetime analysis and proposed two conformers decaying with different kinetics.4 However, the distinctly different decay patterns recorded by us at pHs 7, 4.5 and 1.5 were not observed by them, probably due to the very high concentration of the dye used in their experiments. For the dicationic dye at pH 4.5, Adhikary et al. have reported a longer lifetime component of 3.5 ns for a conformer, which arises when both the benzimidazole units are in planar conformation.23 Apart from a planar structure, Guan et al. also reported multiple binding modes for the dicationic H33258 to DNA, which add another dimension in the mechanistic aspect.22 At pH 7, for H33258, the planar conformation is less pronounced and most of the excited state energy is released via rotation along the benzimide axis. At the same time, the decay behavior observed at pH 11 can be comfortably assigned to a more likely quinonoid structure of the dye, having low emission yield and faster decay processes.4 It is to be mentioned here that there are several instances where the pKa value of the dye in the presence of biomolecules or macrocyclic hosts shows upward or downward shifts depending on the relative binding affinities of the concerned prototropic forms towards the macromolecules.28,30–33 In this case also the presence of negative phosphate backbone may influence in stabilizing certain cationic structure and it is quite likely that the dye pKa may get altered. The inset of Fig. 2 shows the traces recorded for H33258 from a solution at pH 4 and from a solution at pH 7 in presence of CT-DNA. The close similarity among these two decay profiles is notable.
From the fact that the long lifetime, which was observed on binding to DNA/reverse micelles etc. is seen even in aqueous solution of the dye on protonation, it is clear that the structural/orientation changes on protonation of the dye is the key factor for the observed fluorescence changes. In line with the existing reports, it is believed that for monocationic dye significant structural changes occur on protonation and the photoexcited H33258 molecule is believed to undergo fast torsional motion contributing to the non-radiative relaxation to a large extent. Any hinderances that restrict such intramolecular motions, such as structural orientation or restricted medium, will reduce the non-radiative channel. It is clear that the peculiarity came from the dicationic structure in solution at pH 4.5, which displayed nearly single exponential decay with a time constant 3.8 ns and is in agreement with that reported by several others.22,23 Thus, it is reasonable to believe that the intramolecular interactions and the electron density distributions in the dicationic dye force the dye in its planar state, which otherwise is attained by the monocationic form on groove binding on DNA. To understand the ground state structural changes brought out by the successive addition of proton at the nitrogen atoms corresponding to mono-, di-, tricationic forms, the ground state structures were optimized at PM3 level incorporating molecular mechanics (MM) correction using Gaussian 92 package.29 Since the geometry optimization were done without any solvent consideration, these models provide only a qualitative picture of the structures in the ground state.
Fig. 4 shows the optimized ground state structures for the monocationic (I), dicationic (II), tricationic (III) and the anionic (IV) species. A close look at these structures reveals that at pH 7 (I), the three groups (as indicated in the structure) displayed a completely non-planar structure with the two benzimidazole groups positioned at a dihedral angle of 38° and also the phenolic ring flipping out from the plane by 31°. In the dicationic structure, the piperazine substituted benzimidazolium nitrogen gets protonated. This maintains the dye in more planar structure with the benzimidazole groups at 23° and the phenol subunit in plane. Further, the tri-cationic structure with three cationic charges, though the phenolic benzene is in plane, both the benzimidazolium groups, displayed more distortion with a dihedral angle of ∼70°. At pH 11, again the planarity is lost with the benzene ring out by 7°, where as the benzimidazole rings position at a dihedral angle 38°.
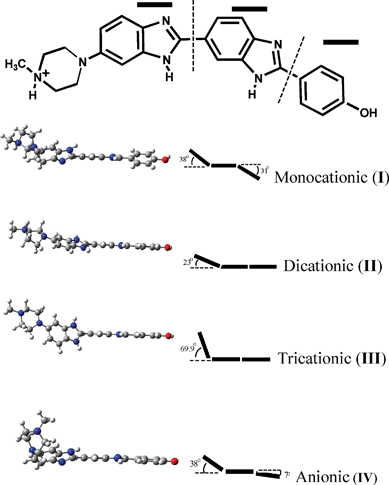 | ||
| Fig. 4 The geometry optimized ground state structures for the different prototopic forms of H33258. The outlined angle represents the out of plane twist angles among the three groups. | ||
On the other hand, if the fast deactivation is due to a flipping motion which destroys the planarity, then one would expect strong temperature dependence on the decay kinetics at pH 7. Also there should be a clear differentiation at pH 4.5, where planarity is maintained. Fig. 5 represents the decay profiles recorded from solutions at pH 7 at different temperatures from 10 °C to 70 °C. In a separate absorption measurement, it has been confirmed that there is no major change in the protonation equilibrium on varying the temperature within the range attempted here. Interestingly, the fluorescence decay at pH 7 responded to the temperature changes in an unprecedented way, which showed an increase in the contribution of faster decay component (∼500 ps) at lower temperature, whereas the slower component, which is ∼3.8 ns, contributed more at higher temperatures. Generally, an increase in temperature would facilitate the flip motion, thus should increase its contribution, however the reverse is observed in this case. From a detailed analysis of these traces, the plot of weighted contributions of the decay constants with temperature displayed gradual decrease in the weight of one of the faster components while the weight of the slower component correspondingly increased, maintaining the ∼100 ps component nearly unaltered, as shown in the inset of Fig. 5. Such an interconversion of the excited states in H33258 with temperature is interesting and measurements were repeated with the solution at pH 4.5 also. As evident from Fig. 6, the decay at pH 4.5 did not show any appreciable change in the decay profile with temperature, giving clear indication about the difference in the relaxation mechanism at pH 7 and 4.5. The temperature dependence on the decay at pH 1.5 showed similar trend as that of pH 7, however, changes are relatively less prominent in the former case. These observations are quite unusual, and certainly point to the direct link between the structural orientation and relaxation mechanism. The interconversion of the faster and slower decay components with temperature can arise in case of a nearly isoenergetic double well potential in the excited state, which are separated by a very small energy barrier as represented schematically in Fig. 7. The shape and depth of the potential surface is determined by the structure of the protonated species of H33258. It is likely that the excited surface of I can be represented by curve a in Fig. 7. Here, excitation of H33258 at pH 7 populates the locally excited state (well A) having a lifetime τ ∼ 400–500 ps. The other minimum (well B) represents a more planar form of the dye, having longer lifetime, about 3.8 ns. As the ambient temperature increases, cross over from well A to well B is facilitated which leads to an increase in the contribution of the 3.8 ns component. On the other hand, lowering the temperature retains the molecule in well A and thus the decay traces display an increase in the contribution from the faster component. Surely, this cross-over could be assigned to the flipping motion between the two benzimidazolium groups, thus bringing the molecule to a more planar conformation. At pH 4.5 for the dicationic dye, the excited state may constitute only well B (curve b) and majority of the populations are retained there and hence does not show any temperature dependence. As indicated from the double bond character between the two benzimidazole goups, this planarity could also represent a slight charge transfer character as claimed in many earlier reports.18
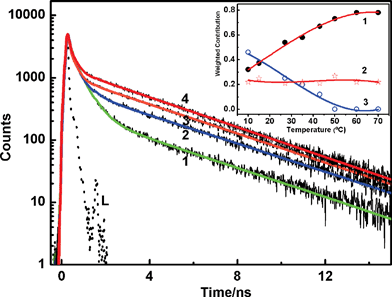 | ||
| Fig. 5 Fluorescence decay traces recorded from H33258 solution at pH 7 at different solution temperatures; 10 °C (1), 35 °C (2), 50 °C (3) and 70 °C (4). L represents the excitation lamp profile. Inset shows the weighted contribution changes for the three decay constants τ1 ∼ 3.6–3.8 ns (1), τ2 ∼ 100–120 ps (2) and τ3 ∼ 400–500 ps (3). | ||
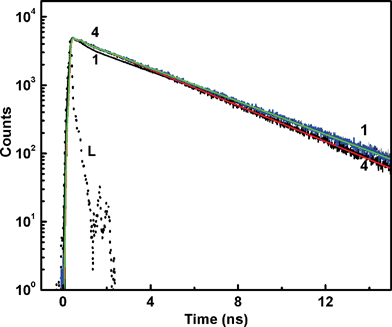 | ||
| Fig. 6 Fluorescence decay traces recorded from H33258 solution at pH 4.5 at different solution temperatures; 10 °C (1), 35 °C (2), 50 °C (3) and 70 °C (4). L represents the excitation lamp profile. | ||
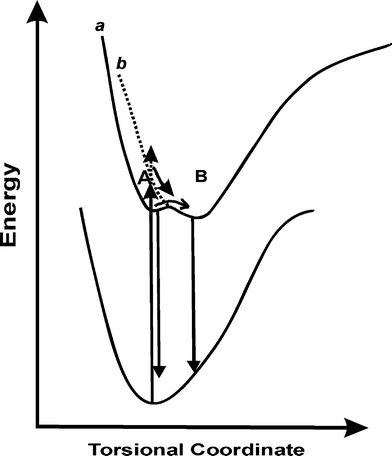 | ||
| Fig. 7 Schematic representation of the excited state dynamics of H33258 at pH 7 (a) and 4.5 (b). | ||
Conclusion
In summary, the intriguing photophysical behavior of H33258 with pH has been studied by steady-state and time-resolved fluorescence measurements. The notable structural differences among its multi-protonated structures are well projected in their emission behavior with pH. It has been seen that a change in the solution pH from 7 to 4.5, enhances the emission yield by ∼20-fold and this change is ∼80-fold on changing the pH from 1.5 to 4.5. While a fast flipping motion among the two benzimidazole rings is considered as the most probable reason for the fast fluorescence decay, a more planar structure of the dicationic dye having a double bond character between the imidazolium groups is the most likely fluorescing species. A similar planar structure is considered as the fluorescent emitting species of H33258 on minor groove binding to DNA. On the basis of temperature dependent fluorescence decay dynamics explored at pH 7 and pH 4, we could suggest that a nearly isoenergetic double well excited state potential is possibly involved with the dye at pH 7. At higher temperature, the conversion to the planar structure is facilitated, enhancing the emission probability. For the dicationic dye at pH 4.5, the emissive species is the planar dye, having a double bond character between the two benzimidazole groups. Due to the many protonation/deprotonation sites in the molecule, the photophysical features of H33258 should be analysed with a clear idea of different protolytic equilibria and the structural orientations feasible at the specified solution condition and is very challenging. Further studies are being carried out to introduce rigid environments for the dye so as to compare its interaction with biomolecules and to draw possible applications in drug binding and release.References
- J. R. Lakowicz, Principles of fluorescence spectroscopy, Springer: New York, 2006 Search PubMed.
- H. LeVine III, Quantification of beta-sheet amyloid fibril structures with thioflavin T, Methods Enzymol., 1999, 309, 274 CAS.
- (a) H. Naiki, K. Higuchi, M. Hosokawa and T. Takeda, Fluorometric determination of amyloid fibrils in vitro using the fluorescent dye, thioflavine T, Anal. Biochem., 1989, 177, 244–249 CAS; (b) C. E. Kung and J. K. Reed, Fluorescent molecular rotors: a new class of probes for tubulin structure and assembly, Biochemistry, 1989, 28, 6678–6686 CrossRef CAS.
- G. Cosa, K.-S. Focsaneanu, J. R. N. McLean, J. P. McNamee and J. C. Scaiano, Photophysical properties of fluorescenct DNA-dyes bound to single and double-stranded DNA in aqueous buffered solution, Photochem. Photobiol., 2001, 73, 585–599 CAS.
- A. C. Bhasikuttan, J. Mohanty, W. M. Nau and H. Pal, Efficient fluorescence enhancement and cooperative binding of an organic dye in a supra-biomolecular host-protein assembly, Angew. Chem., Int. Ed., 2007, 46, 4120–4122 CrossRef CAS.
- A. C. Bhasikuttan, J. Mohanty and H. Pal, Interaction of Malachite Green with Guanine-Rich Single Strand DNA: Preferential Binding to G-Quadruplex, Angew. Chem., Int. Ed., 2007, 46, 9305–9307 CrossRef CAS.
- S. Dutta Choudhury, J. Mohanty, H. Pal and A. C. Bhasikuttan, Cooperative Metal Ion Binding to a Cucurbit[7]uril-Thioflavin T Complex: Demonstration of a Stimulus Responsive Fluorescent Supramolecular Capsule, J. Am. Chem. Soc., 2010, 132, 1395–1401 CrossRef CAS.
- F. G. Loontiens, P. Regenfuss, A. Zechel, L. Dumortier and R. M. Clegg, Binding characteristics of Hoechst 33258 with calf thymus DNA, Poly[d(A-T)], and d(CCGGAATTCCGG): Multiple stoichiometries and determination of tight binding with a wide spectrum of site affinities, Biochemistry, 1990, 29, 9029–9039 CrossRef CAS.
- J.-H. Moon, S. K. Kim, U. Sehlstedt, A. Rodger and B. Norden, DNA structural features responsible for sequence dependent binding geometries of Hoechst 33258, Biopolymers, 1996, 38, 593–606 CrossRef CAS.
- L. Goracci, R. Germani, G. Savelli and D. M. Bassani, Hoechst 33258 as a pH-sensitive probe to study the interaction of amine oxide surfactants with DNA, ChemBioChem., 2005, 6, 197–203 CrossRef CAS.
- H. Ojha, B. M. Murari, S. Anand, M. I. Hassan, F. Ahmad and N. K. Chaudhury, Interaction of DNA minor groove binder Hoechst 33258 with Bovine Serum Albumin, Chem. Pharm. Bull., 2009, 57, 481–486 CrossRef CAS.
- N. V. Lyubimova, P. G. Coultas, K. Yuen and R. F. Martin, In vivo radioprotection of mouse brain endothelial cells by Hoechst 33342, Br. J. Radiol., 2001, 74, 77–82 Search PubMed.
- L. Denison, A. Haigh, G. D'Cunha and R. F. Martin, DNA Ligands as Radioprotectors: Molecular Studies with Hoechst 33342 and Hoechst 33258, Int. J. Radiat. Biol., 1992, 61, 69–74 CrossRef CAS.
- T. R. Downs and W. W. Wilfinger, Fluorometric quantification of DNA in cells and tissues, Anal. Biochem., 1983, 131, 538–547 CAS.
- C. F. Cesarone, C. Bolognesi and L. Santi, Improved microfluorometric DNA determination in biological material using 33258 Hoechst, Anal. Biochem., 1979, 100, 188–197 CAS.
- D. L. Stout and F. F. Becker, Fluorometric quantitation of single-stranded DNA: A method applicable to the technique of alkaline elution, Anal. Biochem., 1982, 127, 302–307 CrossRef CAS.
- M. Ladinig, W. Leupin, M. Meuwly, M. Respondek, J. Wirz and V. Zoete, Protonation equilibria of Hoechst-33258 in aqueous solution, Helv. Chim. Acta, 2005, 88, 53–67 CrossRef CAS.
- K. Kalninsh, D. V. Pestov and Y. K. Roshchina, Absorption and fluorescence spectra of the probe Hoechst 33258, J. Photochem. Photobiol., A, 1994, 83, 39–47 CrossRef CAS.
- T. Stokke and H. B. Steen, Multiple binding modes for Hoechst 33258 to DNA, J. Histochem. Cytochem., 1985, 33, 333–338 CAS.
- H. Gorner, Direct and sensitized photoprocesses of bis-benzimidazole dyes and the effects of surfactants and DNA, Photochem. Photobiol., 2001, 73, 339–348 CrossRef CAS.
- M. Rahimian, Y. Miao and W. D. Wilson, Influence of DNA structure on adjacent site cooperative binding, J. Phys. Chem. B, 2008, 112, 8770–8778 CrossRef CAS.
- Y. Guan, R. Shi, X. Li, M. Zhao and Y. Li, Multiple binding modes for dicationic Hoechst 33258 to DNA, J. Phys. Chem. B, 2007, 111, 7336–7344 CrossRef CAS.
- A. Adhikary, V. Buschmann, C. Muller and M. Sauer, Ensemble and single-molecule fluorescence spectroscopic study of binding modes of the bis-benzimidazole derivative Hoechst 33258 with DNA, Nucleic Acids Res., 2003, 31, 2178–2186 CrossRef CAS.
- S. K. Pal, L. Zhao and A. H. Zewail, Water at DNA surfaces: Ultrafast dynamics in minor groove recognition, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 8113–8118 CrossRef CAS.
- D. Banerjee and S. K. Pal, Ultrafast charge transfer and solvation of DNA minor groove binder: Hoechst 33258 in restricted environments, Chem. Phys. Lett., 2006, 432, 257–262 CrossRef CAS.
- V. N. Umetskaya and Y. M. Rozanov, Mechanism of the interaction of DNA with the fluorescent dye Hoechst 33258, Biophysika, 1990, 35, 399–401 Search PubMed.
- K. E. Furse and S. A. Corcelli, The dynamics of water at DNA interfaces: computational studies of Hoechst 33258 bound to DNA, J. Am. Chem. Soc., 2008, 130, 13103–13109 CrossRef CAS.
- M. Shaikh, J. Mohanty, A. C. Bhasikuttan, V. D. Uzunova, W. M. Nau and H. Pal, Salt-induced Guest Relocation from a Supramolecular Cavity into a Biomolecular Pocket: Interplay between Cucurbit[7]uril and Albumin, Chem. Commun., 2008, 3681–3683 RSC.
- M. J Frisch, G. W. Trucks, M. Head-Gorden, P. M. W. Gill, M. W. Wong, J. B. Foresman, B. G. Johnson, H. B. Schlegel, M. A. Robb, E. S. Replogle, R. Gomperts, J. L. Andres, K. Rahavachari, J. S. Binkley, C. Gonzalez, R. Martin, L. D. J. Fox, D. J. Defrees, J. Baker, J. J. P. Stewart, J. A. Pople, Gaussian 92, Gaussian, Inc, Pittsburgh, PA, 1992 Search PubMed.
- J. F. G. Walz, B. Terenna and D. Rolinee, Equilibrium studies on neutral red-DNA binding, Biopolymers, 1975, 14, 825–837 CrossRef.
- J. Mohanty, A. C. Bhasikuttan, W. M. Nau and H. Pal, Host–guest complexation of neutral red with macrocyclic host molecules: Contrasting pKa shifts and binding affinities for cucurbit[7] uril and β-cyclodextrin, J. Phys. Chem. B, 2006, 110, 5132–5138 CrossRef CAS.
- M. Shaikh, J. Mohanty, P. K. Singh, W. M. Nau and H. Pal, Complexation of Acridine Orange by Cucurbit[7]uril and beta-cyclodextrin: Photophysical Effects and pKa Shifts, Photochem. Photobiol. Sci., 2008, 7, 408–414 RSC.
- R. L. Jones and W. D. Wilson, Effect of ionic strength on the pKa of ligands bound to DNA, Biopolymers, 1981, 20, 141–154 CrossRef CAS.
| This journal is © The Royal Society of Chemistry and Owner Societies 2011 |