Photocatalytic discoloration of aqueous malachite green solutions by UV-illuminated TiO2 nanoparticles under air and nitrogen atmospheres: effects of counter-ions and pH
Julián Andrés
Rengifo-Herrera
*a,
Luis René
Pizzio
a,
Mirta Noemí
Blanco
a,
Christophe
Roussel
*cd and
César
Pulgarin
b
aCentro de Investigación y Desarrollo en Ciencias Aplicadas “Dr J. J. Ronco” (CINDECA), Departamento de Química, Facultad de Ciencias Exactas, UNLP-CCT La Plata, CONICET, 47 No. 257, 1900 La Plata, Buenos Aires, Argentina. E-mail: julianregifo@quimica.unlp.edu.ar; Fax: +54 221 4211353 ext.125; Tel: +54 221 421353
bSB-ISIC-GGEC, Station 6, Ecole Polytechnique Fédérale de Lausanne, CH-1015, Lausanne, Switzerland
cSB-SCGC, Station 6, Ecole Polytechnique Fédéral de Lausanne, CH-1015, Lausanne, Switzerland
dSB-ISIC, Station 6, Ecole Polytechnique Fédéral de Lausanne, CH-1015, Lausanne, Switzerland
First published on 26th October 2010
Abstract
Under air atmosphere, the photocatalytic discoloration of malachite green (MG) aqueous solutions (a triphenylmethane dye) in the presence of TiO2 and UV light followed an oxidative pathway, involving an N-demethylation process evidenced by a blue shifting of the main absorption band with a maximum at 618 nm. This oxidative process was affected by the nature of the dye counter-ion and the pH of the solution. At pH 6.0, the oxidation was found to be faster than at pH 3.0, perhaps due to the poor interactions between MG and the semiconductor surface. Furthermore, with the presence of oxalate as counter-ion, the oxidative photocatalytic discoloration was negatively affected mainly at acidic pH. Under nitrogen atmosphere, some evidence was found about the double behaviour of MG when involved in the photocatalytic discoloration reactions pertaining to TiO2 under these conditions. MG could be simultaneously oxidized, forming N-demethlyated by-products, or reduced, thus leading to leuco-malachite green (LMG) (a colorless and toxic substance) as the main product. The LMG formation is favoured at low pH in the presence of oxalate as counter-ion.
1. Introduction
In the last 25 years, heterogeneous photocatalysis over TiO2 has generated growing interest as a promising technology to degrade chemical substances and inactivate pathogen cells in aqueous solution, since titanium dioxide presents suitable features such as low cost and ready commercial availability.1,2This process consists in the generation of conduction band electron and valence band hole pair (e−CB/h+VB) on TiO2 under UV illumination. These charge carriers can migrate to the semiconductor surface and thus react with suitable electron donors and acceptors producing oxidative and reductive reactions. This photo-induced process leads to the generation of reactive oxygen species (ROS) with high oxidative power such as the hydroxyl radical (˙OH).3–5 This process was used to oxidize organic pollutants of water.6 The photo-induced process could be summarized as follows:
| TiO2 + hνUV → TiO2(e−CB + h+VB) | (1) |
| TiO2(h+VB) + H2O(ads) → ˙OH(ads) + H+ + TiO2 | (2) |
| TiO2(e−CB) + O2 → O2−˙ + TiO2 | (3) |
| O2−˙ + H+ → HO2˙ (pK = 4.5) | (4) |
Heterogeneous photocatalysis over TiO2 has often been used to bleach and degrade aqueous solutions of organic dyes7 or, more recently, biomolecules such as DNA with the aim of designing biocaptors.8
Most of these studies have reported an oxidative photocatalytic discoloration of dye aqueous solutions.9–12 However, some authors13,14 have pointed out that in the particular case of methylene blue (MB), it is possible that under acidic conditions and N2 atmospheres, or even under the presence of molecular oxygen, the photocatalytic discoloration of MB solutions is carried out by a reductive pathway involving the reaction of MB with photo-produced e−CB of the TiO2. This reductive reaction promotes the formation of the doubly reduced form, leuco methylene blue, a colorless substance stabilized at low pH.
Malachite green (MG) is a triphenylmethane dye often used in the aquaculture industry to control fungal and protozoan infections of fishes.15 MG can be easily absorbed in fishs' tissues and then in situ converted into leuco-malachite green (LMG) as the main metabolic by-product.16 Both MG and LMG (Fig. 1) are substances considered to have a high teratogenic potential and environmental persistence.17
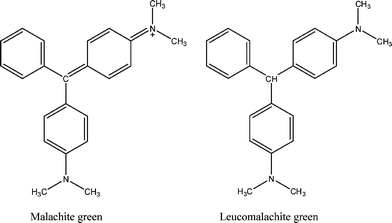 | ||
| Fig. 1 Chemical structures of malachite green (MG) and leuco-malachite green (LMG). | ||
It is well known that MG solutions can undergo photochemical oxidative and reductive fading.18,19 The photochemical pathway of fading oxidation, favoured under aerobic conditions, can induce demethylation of N–Me dyes or the cleavage of the central Ar–C bond, thus leading to amino-substituted benzophenones. The reductive pathway generates LMG, the latest being favoured under anoxic conditions and in the presence of a proton donor.
Only three studies reporting the oxidation of MG on TiO2 surfaces under UV irradiation have been published,20–22 the most interesting for us is the one by Chen et al.22 In this paper, they have demonstrated that under oxic conditions, MG can lead to an oxidative degradation on TiO2 surfaces thus producing N-demethylated by-products during the first steps of the reaction. This oxidation is promoted at neutral pH (7.0) and basic pH (9.0). Under acidic conditions (pH 3.0), MG aqueous solutions are photocatalytically bleached slowly, because under these conditions TiO2 surfaces are positively charged avoiding the interaction and adsorption of MG.
To the best of our knowledge, the influences of the counter ion of the dye onto the heterogeneous photocatalytic processes on TiO2 are rarely explored. In particular, there is no study on the effect of the counter ions on the photocatalytic discoloration of MG aqueous solutions and its reduction on TiO2 nanoparticles. This is an important issue, since it is necessary to know if the photocatalytic discoloration of MG aqueous solutions can also lead reductive processes on TiO2 surfaces then generating its toxic leuco form.
In the present work, the UV induced photocatalytic reactions of MG oxalate (MG-oxalate/TiO2/UV) and MG chloride (MG-Cl−/TiO2/UV) on TiO2 Degussa P-25 were studied under air and nitrogen conditions at two pH values: 3.0 and 6.0.
2. Experimental
2.1 Materials
A commercial powder of TiO2 Degussa P-25 (70% anatase and 30% rutile with a specific surface area around 50 g m−2 was obtained from Degussa AG and used as received. Malachite green oxalate (dye content ≥ 90%) and malachite green chloride (pKa = 6.9)15 (dye content ≥ 96%) were purchased from Sigma–Aldrich and Fluka respectively and used without further purification.HPLC-grade acetonitrile was purchased from Fisher Scientist and double-distilled Milli-Q water was used throughout this study.
2.2 Photocatalytic experiments under air (MG and nitrogen atmospheres)
TiO2 (1.0 g L−1 this concentration showed the best photocatalytic activity towards MG discoloration under the experimental conditions used (data not shown) was added to malachite green aqueous solutions containing 5.0 × 10−4 M either oxalate or chloride into the cylindrical Pyrex bottles (50 mL). Prior to UV irradiation, the resulting suspensions were kept under magnetic stirring in the dark for ca. 30 min to ensure that adsorption/desorption processes dye/TiO2 surface be reached. The solutions were then irradiated by 5 black light lamps Phillips TLD 18 W (emission spectra in the range of 330–400 nm) (Fig. 2), UV intensity between 300–400 nm was around 38 W m−2 and was monitored with a Kipp & Zonen (CM3) power meter (Omni instruments Ltd, Dundee, UK). Atmosphere into the cylindrical Pyrex bottles was controlled by continuous bubbling of either natural oxygen (present in the air) or nitrogen (99.999% Carbagas Switzerland) guaranteeing saturated oxygen and nitrogen conditions. Samples were taken at different illumination times filtered through 0.22 μm membranes of pore size and the discoloration of MG solutions were followed by UV-vis photo-spectrometry (Varian Cary 1-E) monitoring the absorbance at 618 nm. The initial pH of each solution was tuned and adjusted by adding either HCl or NaOH. pHs >6.0 were not tested because the instability of MG at pHs above to its pKa. The running temperature was never higher than 38 °C. | ||
| Fig. 2 Experimental set up used during the photocatalytic experiments carried out under air and N2 saturated atmospheres. | ||
HPLC analyses were carried out on a Hewlett–Packard series 1100 apparatus equipped with a reverse phase silica column (Phenomenex Luna 5 μ C-18). The HPLC analyses were run at 1.0 mL min−1, using an isocratic mode with a mobile phase made of acetonitrile–water (10% acetic acid) at 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :60 ratio. A diode array detector (DAD) was used to monitor malachite green and leuco-malachite green at 618 and 260 nm, respectively. Their absorption spectra were also recorded. The given results are expressed by the average of three experimental runs taking in account the spectrophotometric analyses (standard deviations were found to be equal or lower than 8% for spectrophotometric analyses)
:60 ratio. A diode array detector (DAD) was used to monitor malachite green and leuco-malachite green at 618 and 260 nm, respectively. Their absorption spectra were also recorded. The given results are expressed by the average of three experimental runs taking in account the spectrophotometric analyses (standard deviations were found to be equal or lower than 8% for spectrophotometric analyses)
3. Results and discussion
3.1 Photocatalytic discoloration of malachite green solutions under air atmosphere.
Fig. 3a and 3b show the discoloration of an aqueous solution containing 5.0 × 10−4 M of MG oxalate (MG-oxalate/TiO2/UV) and chloride (MG-Cl−/TiO2/UV) respectively in the presence of TiO2 (1.0 g L−1) under UV irradiation at pH 3.0 and 6.0. Control experiments ran in dark conditions revealed that in both systems (MG-oxalate/TiO2 and MG-Cl−/TiO2) the adsorption of MG on TiO2 was higher at pH 6.0 than at pH 3.0. UV light irradiation in absence of TiO2 did not induce any changes on the MG absorption spectra.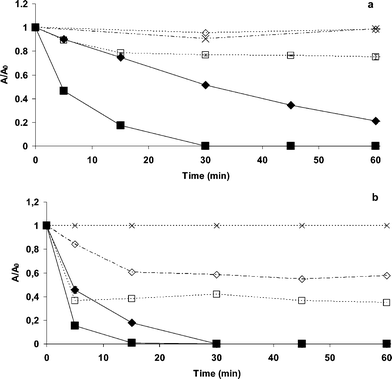 | ||
| Fig. 3 UV-induced photocatalytic discoloration of MG aqueous solutions: oxalate (a) and chloride (b) carried out under oxic conditions in presence of TiO2. (♦) at pH 3.0, (■) at pH 6.0, (◇) dark experiment at pH 3.0, (□) dark experiment at pH 6.0, (×) MG solution exposed under UV light in absence of TiO2. | ||
When MG solutions containing TiO2 nanoparticles were exposed under UV light, discoloration was observed in systems MG-oxalate/TiO2/UV and MG-Cl−/TiO2/UV whatever the pH under study. Moreover, based on Fig. 3, is evident that pH plays a key role on the photocatalytic discoloration of MG aqueous solutions since this reaction proceeds faster at pH 6.0 than at pH 3.0. However, on the other hand, there is also an effect caused by the counter ion nature; Fig. 3 reveals also in system MG-oxalate/TiO2/UV that the photocatalytic discoloration was negatively affected by oxalate presence being this effect more pronounced at pH 3.0.
Photocatalytic discoloration under air atmosphere seems to be oxidative. When the UV-Vis spectra of systems MG-oxalate/TiO2/UV and MG-Cl−/TiO2/UV are considered (Fig. 4a and 4b); blue shifting (hypsochromic) of the main absorption band of MG with maximum at 618 nm is observed whatever the pH.
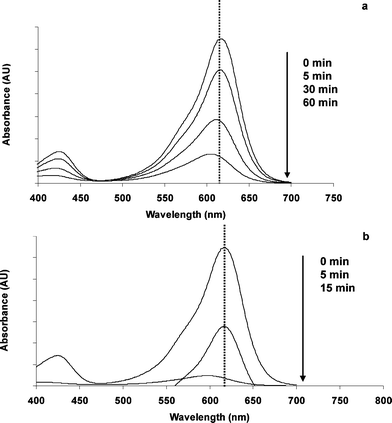 | ||
| Fig. 4 UV-Vis spectra of MG-oxalate in aqueous solution during UV-irradiation and the presence of TiO2 under oxic conditions at (a) pH 3.0 and (b) pH 6.0. | ||
This hypsochromic shifting could be due to the formation of oxidized N-demethylated substances such as monode-MG (λmax = 605 nm) and/or dides-MG symmetrical (λmax = 594 nm).16 These substances could be generated by oxidation, either directly by the photo-induced valence band holes or indirectly by ˙OH radicals.
This reaction between MG and TiO2 was already reported by Chen et al.22 but only at pH 7.0 and 9.0. At pH 3.0, they claim that this effect was not observed. This was explained by the fact that at pH 3.0, the positively charged MG is repelled by the TiO2 surface (also positively charged, pHzpc = 6.023) thus avoiding its adsorption. The photo-degradation could then only be achieved by ˙OH radicals able to migrate from the TiO2 surface towards some distance at the bulk solution as was recently suggested by Enriquez et al.,24,25 thus destroying the whole conjugated chromophore structure. At TiO2 pHzpc, the MG can be adsorbed by hydrogen bonding interactions (physisorption) with the titanol groups (Ti–OH) present at the TiO2 surface.
However, it is not possible to mention that at pH 3.0, the entire TiO2 surface is positively charged. Kormann et al.23 demonstrated by theoretical calculations that at pH 3.0 some Ti–OH groups are also present. Indeed, a lower adsorption of MG through hydrogen bonding interactions results in a slower oxidation than at pH 6.0. Moreover, it was also suggested that basic substrates can interact with acidic TiIV sites.26,27 According to the chemical structure of MG (Fig. 1), adsorption on acidic TiIV thus resulting in TiIV–N(CH3)2 interaction could not be neglected. All the mechanisms cited above could explain the hypsochromic shifting caused by N-demethylation of MG at acidic pH since some MG molecules can weakly interact with the TiO2 surface and thus can be exposed to the attack of oxidative species produced by the photocatalytic process.
In addition, at pH 3.0, in the system MG-oxalate/TiO2/UV, the presence of oxalate produced a detrimental effect on the photo-induced reaction.
Actually, oxalate can interact strongly with TiO2 surfaces at pH < pHzpc by forming inner-sphere complexes, i.e. chemisorptions.28 During the photocatalytic process, organic substances chemisorbed on TiO2 can react rapidly with shallowly trapped holes while weakly adsorbed molecules, i.e. physisorbed, can react with deeply trapped holes, the latter being a slower process.29 As a consequence, MG weakly adsorbed cannot react efficiently with the photo-induced oxidative species (h+, ˙OH). This effect is not observed for the system MG-Cl−/TiO2/UV since Cl− does not compete with MG by valence band holes or ˙OH radicals as already pointed out by Guillard et al.30 when studying the photocatalytic discoloration of methylene blue on TiO2. This demonstrates that MG oxidation on TiO2 is a process where the interactions between dye and the photocatalyst surface play an important role. At pH 6.0, the adsorption of oxalate is reduced27 favouring a better interaction between MG and the TiO2 surface, thus, enhancing the photocatalytic discoloration.
3.2 Photocatalytic discoloration of malachite green solutions under nitrogen atmospheres
When N2 was continuously bubbled in the following systems: MG-oxalate/TiO2/UV and MG-Cl−/TiO2/UV, four interesting facts could be pointed out (Fig. 5a and b): (i) MG adsorption in dark conditions for the system MG-oxalate at pH 3.0 is enhanced; (ii) there is a much faster photocatalytic discoloration mainly for the system MG-oxalate/TiO2/UV at pH 3.0 compared to oxic conditions (at the same pH); (iii) for all the systems, the photocatalytic discoloration at pH 6.0 was faster than it was already observed under air atmosphere (Fig. 3); (iv) in the system MG-Cl−/TiO2/UV, the solution discoloration was slow compared to the system MG-oxalate/TiO2/UV (Fig. 5a and b).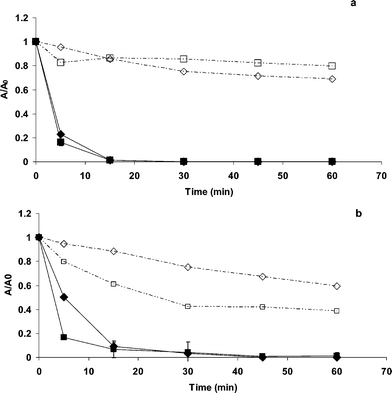 | ||
| Fig. 5 UV-induced photocatalytic discoloration of MG aqueous solutions: oxalate (a) and chloride (b) carried out under anoxic conditions in presence of TiO2. (♦) at pH 3.0, (■) at pH 6.0, (◇) dark experiment at pH 3.0, (□) dark experiment at pH 6.0. | ||
According to the UV-Vis spectra depicted on Fig. 6a, the previously reported blue shifting for the system MG-oxalate/TiO2/UV, attributed to the N-demethylation of MG (induced by oxidation, Fig. 4a) was not observed. In contrast, the system MG-Cl−/TiO2/UV gave evidence of a slight blue shifting (at 605 nm) but, only after 15 min of UV irradiation (Fig. 6b). Upon these observations, two questions arise. How is it possible that under N2 atmosphere, where there is no electron acceptor able to avoid the charge carrier recombination, the photocatalytic discoloration of aqueous MG solutions can take place? Why was the adsorption of MG enhanced in the system MG-oxalate/TiO2/UV at pH 3.0?
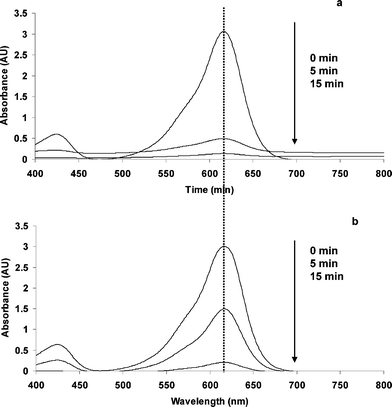 | ||
| Fig. 6 UV-Vis spectral changes induced by UV-irradiation of (a) MG-oxalate and (b) MG-chloride aqueous solutions in the presence of TiO2 under N2 atmosphere at pH 3.0. | ||
One of the possible explanations to the first question could stem from the participation of MG as donor and/or acceptor of electrons during the photocatalytic reaction. Furthermore, the answer to the second question makes it clear that oxygen could compete with MG for certain adsorption sites on TiO2. There are some reports in the literature5 claiming that reduction and oxidation on TiO2 nanoparticles is achieved on different sites or crystalline faces. Thus, when oxygen is absent, reductive sites might remain free and MG could be easily adsorbed and then reduced.
Under saturated N2 atmosphere, the participation of MG as electron donor or acceptor could be evidenced when following the photocatalytic reaction by HPLC. For instance, for the system MG-oxalate/TiO2/UV at pH 3.0, HPLC analysis (recorded at 618 nm) at the beginning of the reaction revealed only one peak at a retention time of 1.5 min corresponding to MG (Fig. 7a). After 60 min under UV irradiation a new peak with a retention time of 1.3 min was also observed (Fig. 7b). Deeper UV-Vis analysis revealed that the maximum of absorbance of this product is 605 nm. This new peak could correspond to an N-demethylated by-product even if the UV-Vis spectra depicted in Fig. 6a did not reveal it clearly and thus pointing out the possible role of MG as electron donor in place of O2. In addition, when recorded HPLC measurements were done at 256 nm, a new peak corresponding to a more hydrophobic compound could be observed at a retention time of 4.4 min after 1 h of UV irradiation of GM-oxalate/TiO2/UV at pH 3.0 (Fig. 8). The UV-Vis spectrum of this product is rather similar to that of leuco-malachite green LMG as reported by Cho et al.16HPLC analyses performed for the systems MG-oxalate/TiO2/N2/UV at pH 6.0 and MG-Cl−/N2/UV either at pH 3.0 or 6.0 did not show this particular peak, but nevertheless the presence of N-demethylated by-products were also detected (data not shown). These results could point out the dual role of MG in photocatalytic reactions in absence of oxygen. It suggests that this molecule could participate either as electron donor or acceptor. Indeed, in the absence of oxygen, MG could be reduced to LMG (through the radical anion ˙MG−), the photoreduction process was favored in the presence of compounds which can be oxidized faster than MG and/or proton donors.18,19 Since proton donors stabilize MG−˙ radicals, the whole reduction of MG leads directly to LMG in acidic medium.
| e−CB + MG → ˙MG− | (5) |
| ˙MG− + H+ → LMG | (6) |
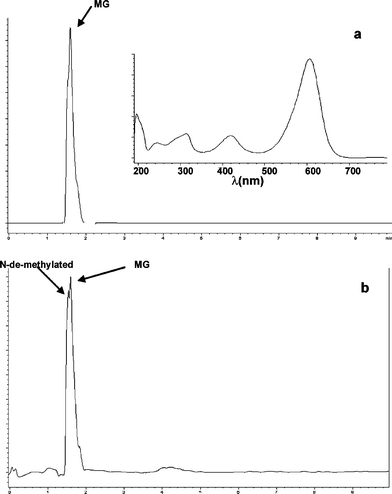 | ||
| Fig. 7 Chromatograms using DAD detector at 620 nm (a) before UV-irradiation and (b) after 1 h of UV-irradiation for the system MG-oxalate/TiO2 under N2 atmosphere at pH 3.0. Insert in Fig. 7a shows the DAD spectrum of chromatographic peak with retention time 1.5 min corresponding to MG. | ||
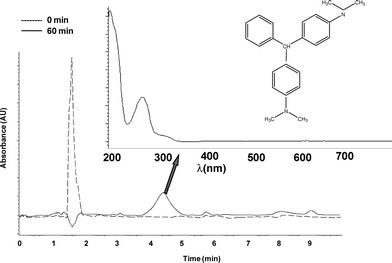 | ||
| Fig. 8 Chromatograms using DAD detector at 256 nm in the system MG-oxalate/TiO2 after 1 h of UV light irradiation under N2 atmosphere at pH 3.0. Insert shows the UV-Vis spectrum of this peak. | ||
It is also important to mention the role of the counter-ion. At pH 3.0 for the system MG-oxalate/TiO2/UV, the oxalate adsorption is favoured, thus leading to an efficient trapping of the photo-generated Valence band holes on the TiO2. In this case, oxalate behaves as a sacrificial electron donor (SED) avoiding the electron recombination, then inducing the reduction of MG to LMG by the photo-excited electrons of the TiO2 conduction band. In contrast, for the systems MG-oxalate/TiO2/UV at pH 6.0 and MG-Cl−/TiO2/UV either at pH 3.0 or 6.0, the formation of LMG would not be favoured. For the system MG-oxalate/TiO2/UV at pH 6.0, oxalate adsorption is reduced within its h+VB scavenging effect. Furthermore, in such pH conditions, the stabilization of MG−˙ radicals by H+ is also minimized preventing from the LMG formation. For the system MG-Cl−/TiO2/UV, the formation of LMG will also be negatively affected as Cl− is not a suitable electron donor.30 In this case, both MG and LMG could be oxidised.
4. Conclusions
The results found in this study might suggest that under air saturated atmospheres the photocatalytic discoloration of systems MG-oxalate/TiO2/UV and MG-Cl−/TiO2/UV was carried out by an oxidative process, where the MG undergoes N-demetylation reactions evidenced by a hypsochromic shifting of its main absorption peak at 618 nm either at pH 3.0 or 6.0. The counter-ion played an important role since when oxalate was present the photocatalytic discoloration was negatively affected, mainly at pH 3.0. This fact could be attributed to the oxalate adsorption competing with the MG and thus leading to the scavenging of the photo-generated holes h+VB. At pH 6.0, whereas MG can interact strongly with the photocatalyst surface, oxalate adsorption is reduced; therefore, MG oxidation would be enhanced in such conditions.Experiments carried out in the systems MG-oxalate/TiO2/UV and MG-Cl−/TiO2/UV under N2 saturated atmospheres might allow suggesting the possible dual role played by MG: either as electron donor or acceptor depending on both the counter-ion nature and the pH of the solution. When oxalate was strongly adsorbed on the TiO2 surface at pH 3.0, the reduction of MG to LMG was favoured since oxalate could act as an efficient sacrificial electron donor (SED) leaving the photo-generated electrons on the TiO2 conduction band available for the reduction of MG. Adsorption of MG was probably enhanced in the absence of oxygen at this pH. At pH 6.0, a stronger interaction between MG and TiO2 surface promotes the oxidation of the dye. Presence of Cl− as counter-ion diminishes the possibilities of MG to be reduced, since this ion is a poorer electron donor than the oxalate anion.
However, further studies must be done in order to confirm the generation of LMG under N2 saturated atmospheres. Furthermore, another key point to be studied would be the role of low oxygen concentrations on the generation of LMG.
Acknowledgements
The authors thank the Cooperation@EPFL for its support to the Argentinean–Swiss cooperation project: “Argentinean–Swiss cooperation action in based TiO2 photo-active nano-materials for bacteria inactivation and pollutant oxidation in solid-gas and solid-water interfaces” and CONICET for the support to the project “Catalizadores heterogéneos coadyuvantes del cuidado del medio ambiente” (No. PIP 1938 2009-2011). J. A. Rengifo-Herrera thanks Dr Walter Torres H. (Universidad del Valle, Cali, Colombia) for the support and collaboration in the beginning of this research.References
- M. R. Hoffmann, S. T. Martin, W. Choi and D. W. Bahnemann, Environmental applications of semiconductor photocatalysis, Chem. Rev., 1995, 95, 69–69 CrossRef CAS.
- A. L. Linsebigler, G. Lu and J. T. Yates, Photocatalysis on TiO2 surfaces: Principles, mechanisms, and selected results, Chem. Rev., 1995, 95, 735–758 CrossRef CAS.
- A. Mills, Davies, R. Worsley and D. Water, Purification By Semiconductor Photocatalysis, Chem. Soc. Rev., 1993, 22, 417–426 RSC.
- A. Fujishima, T. N. Rao and D. A. Tryk, Titanium Dioxide Photocatalysis, J. Photochem. Photobiol., C, 2000, 1, 1–21 CrossRef CAS.
- A. Fujishima, X. Zhang and D. T. Tryk, TiO2 photocatalysis and related surface phenomena, Surf. Sci. Rep., 2008, 63, 515–582 CrossRef CAS.
- J. Kiwi, C. Pulgarin, P. Peringer and M. Graetzel, Beneficial Effects of Heterogeneous Photocatalysis on the Biodegradation of of Anthraquinone Sulfonate Observed in Water Treatment, New J. Chem., 1993, 17, 487 Search PubMed.
- M. A. Rauf and S. Salman Ashraf, Fundamental principles and application of heterogeneous photocatalytic degradation of dyes in solution, Chem. Eng. J., 2009, 151, 10–18 CrossRef CAS.
- J. F. Liu, C. Roussel, G. Lagger, P. Tacchini and H. H Girault, Antioxidant sensors based on DNA-modified electrodes, Anal. Chem., 2005, 77, 7687 CrossRef CAS.
- M. H. Habibi, A. Hassanzadeh and S. Mahdavi, The effect of operational parameters on the photocatalytic degradation of three textile azo dyes in aqueous TiO2 suspensions, J. Photochem. Photobiol., A, 2005, 172, 89–96 CrossRef CAS.
- N. M. Mahmoodi, M. Arami, N. Y. Limaee, K. Gharanjig and F. D. Ardejani, Decoloration and mineralization of textile dyes at solution bulk by heterogeneous nanophotocatalysis using immobilized nanoparticles of titanium dioxide, Colloids Surf., A, 2006, 290, 125–131 CrossRef CAS.
- K. Wang, J. Zhang, L. Lou, S. Yang and Y. Chen, UV or visible light induced photodegradation of AO7 on TiO2 particles: the influence of inorganic ions, J. Photochem. Photobiol., A, 2004, 165, 201–207 CrossRef CAS.
- A. Bianco, C. Baiocchi, M. C. Brussino, P. Savarino, V. Augugliaro, G. Marci and L. Palmisano, Photocatalytic degradation of acid blue 80 in aqueous solutions containing TiO2 suspensions, Environ. Sci. Technol., 2001, 35, 971–976 CrossRef CAS.
- N. R. De Tacconi, J. Carmona and K. Rajeshwar, Reversibility of photoelectrochromism at the TiO2/methylene blue interface, J. Electrochem. Soc., 1997, 144, 2486–2490 CrossRef CAS.
- A. Mills and J. Wang, Photo-bleaching of methylene blue sensitized by TiO2: an ambiguous system?, J. Photochem. Photobiol., A, 1999, 127, 123–134 CrossRef CAS.
- D. J. Alderman, Malachite green: a review, J. Fish Dis., 1985, 8, 289–298 CrossRef CAS.
- B. P. Cho, T. Yang, L. R. Blankenship, J. D. Moody, M. Churchwell, F. A. Beland and S. J. Culp, Synthesis and characterization of N-demethylated metabolites of malachite green and leuco-malachite green, Chem. Res. Toxicol., 2003, 16, 285–294 CrossRef CAS.
- K. V. K. Rao, Inhibition of DNA synthesis in primary rat hepatocytes by malachite green: a new liver tumor promoter, Toxicol. Lett., 1995, 81, 107–113 CrossRef CAS.
- N. Allen, B. Mohajerani and J. Richards, Photofading of basic triphenylmethane dyes: evidence of electron transfer, Dyes Pigm., 1981, 2, 31–35 CrossRef CAS.
- D. Duxburry, The photochemistry and photophysics of triphenylmethane dyes in solid and liquid media, Chem. Rev., 1993, 93, 381–433 CrossRef CAS.
- J. Zhao, K. Wu, T. Wu, H. Hidaka and N. Serpone, Photodegradation of dyes with poor solubility in aqueous surfactant/TiO2 dispersion under visible light irradiation, J. Chem. Soc., Faraday Trans., 1998, 94, 673–676 RSC.
- H. Kominami, H. Kumamoto, Y. Kera and B. Ohtani, Photocatalytic decolorization and mineralization of malachite green in an aqueous suspension of titanium (IV) oxide nano-particles under aerated conditions: correlation between some physical properties and their photocatalytic activity, J. Photochem. Photobiol., A, 2003, 160, 99–104 CrossRef CAS.
- C. C. Chen, C. S. Lu, Y. C. Chung and J. L Jan, UV induced photodegradation of malachite green on TiO2 nanoparticles, J. Hazard. Mater., 2007, 141, 520–528 CrossRef CAS.
- C. Kormann, D. W. Bahnemann and M. R. Hoffmann, Photolysis of chloroform and other organic molecules in aqueous TiO2 suspensions, Environ. Sci. Technol., 1991, 25, 494–500 CAS.
- R. Enriquez, A. G. Agrios and P. Pichat, Probing multiple effects of TiO2 sintering temperature on photocatalytic activity in water by use of a series of organic pollutant molecules, Catal. Today, 2007, 120, 196–202 CrossRef.
- R. Enriquez and P. Pichat, Different net effect of TiO2 sintering temperature on the photocatalytic removal rates of 4-chlorophenol, 4-chlorobenzoic acid, and dichloroacetic acid in water, J. Environ. Sci. Health, A, 2006, 41, 955–966 Search PubMed.
- G. Martra, Lewis acid and base sites at the surface of microcrystalline TiO2 anatase: relationships between surface morphology and chemical behaviour, Appl. Catal., A, 2000, 200, 275–285 CrossRef CAS.
- C. F. Lien, Y. F. Lin, Y.-S. Lin, M.-T. Chen and J.-L. Lin, FTIR study of adsorption and surface reactions of N(CH33 on TiO2, J. Phys. Chem. B, 2005, 109, 10962–10968 CrossRef CAS.
- S. J. Hug and D. W. Bahnemann, Infrared spectra of oxalate, malonate and succinate adsorbed on the aqueous surface of rutile, anatase and lepidocrocite measured with in situ ATR-FTIR, J. Electron Spectrosc. Relat. Phenom., 2006, 150, 208–219 CrossRef CAS.
- W. Xu and D. Raftery, Photocatalytic oxidation of 2-propanol on TiO2 powder and TiO2 monolayer catalysts studied by solid-state NMR, J. Phys. Chem. B, 2001, 105, 4343–4349 CrossRef CAS.
- C. Guillard, E. Puzenat, H. Lachheb, A. Houas and J. M. Herrmann, Why inorganic salts decreases the TiO2 photocatalytic efficiency, Int. J. Photoenergy, 2005, 07, 1–9 CrossRef CAS.
| This journal is © The Royal Society of Chemistry and Owner Societies 2011 |