Metabolic-targeted therapy with dichloroacetate (DCA): a novel treatment strategy to improve the outcome of photodynamic therapy
Mateusz
Kwitniewski
*a,
Johan
Moan
ab and
Asta
Juzeniene
a
aDepartment of Radiation Biology, Institute for Cancer Research, Norwegian Radium Hospital, Oslo University Hospital, Montebello, Oslo, 0310, Norway. E-mail: kwitniewski@gumed.edu.pl
bInstitute of Physics, University of Oslo, Blindern, 0316, Oslo, Norway
First published on 26th October 2010
Abstract
For the first time we present data showing that metabolic targeted therapy with dichloroacetate (DCA) may improve the outcome of photodynamic therapy. This treatment modality can be easily introduced into clinical practice guidelines.
Photodynamic therapy (PDT) involves light activation of photosensitizing drugs, so-called photosensitizers (PS), and is used for treatment of several types of cancerous and non-cancerous conditions. Photosensitizers for PDT usually accumulate in tumor tissues with some selectivity. Thus, malignant and abnormal cells can be destroyed by PDT which acts by producing singlet oxygen and other reactive oxygen species (ROS). PDT is minimally invasive, considered to be free of severe side effects and, where necessary, can be repeated without any risk of overdosing.1,2 However, the efficiency of PDT when used as a monotherapy is often limited to the illuminated area of the primary tumor and generally does not affect deeper localized cancer cells, metastases or cells which are resistant to direct or indirect cell killing mechanisms.
Cancer cells are often characterized by anaerobic glycolysis in the presence of oxygen (Warburg's effect) and a concomitant decrease in mitochondrial respiration, which suppresses apoptosis.3,4 A glycolytic environment is associated with an antiapoptotic and pro-proliferative state, characterizing most solid tumors.5
Dichloroacetate (DCA), a small molecule of 150 Da, is a metabolic modulator that has already been used in humans in the treatment of lactic acidosis and inherited mitochondrial diseases and it is well tolerated by patients.6 It was shown that DCA shifts cancer cell metabolism from glycolysis to glucose oxidation, decreases mitochondrial membrane potential, produces reactive oxygen species, induces apoptosis, decreases proliferation, and may inhibit tumor growth, without affecting normal cells.7,8 It is highly bioavailable and can penetrate the blood-brain barrier. DCA activates the pyruvate dehydrogenase (PDH) by inhibition of pyruvate dehydrogenase kinase 1 (PDK1). Activation of PDH increases glucose oxidation, the entry of pyruvate into the mitochondria and the influx of acetyl-CoA into Krebs cycle, thus increases NADH delivery to the electron transport chain.8 This metabolic reprogramming can increase oxygen consumption within the tumor and, in turn, make the tumor more hypoxic due to the limited oxygen supply caused by the morphological abnormalities of tumor vessels.9,10
Oxygen is crucial for PDT since ROS, mainly singlet oxygen, are generated in the target tissue via energy transfer from the photosensitizer to molecular oxygen. In addition to direct tumor cell kill, microvascular collapse, as frequently observed following PDT, can lead to severe hypoxia which contributes to long-term tumor control.11,12 This suggests that DCA might work well in combination with PDT since the ability of PDT to consume oxygen and to block blood vessels will decrease the amount of oxygen available to tumors and, therefore, make cells in tumors more susceptible to the effects of DCA.
The aim of this study was to determine whether the outcome of 5-aminolevulinic acid (ALA) based PDT of human A431 squamous cell carcinoma and human MCF-7 breast carcinoma cells can be improved when combined with DCA therapy. Normal human fibroblasts F11hTERT were used as a control cell line. Analysis of the influence of DCA on cell viability and the effectiveness of combined treatment were performed.
A431 human squamous carcinoma (ATCC CRL-1555) and MCF-7 human breast cancer cells (ATCC HTB-22) were maintained in DMEM medium (Lonza Group Ltd., Switzerland) supplemented with 2 mM L-glutamine, 100 U ml−1penicillin, 100 μg ml−1streptomycin and 10% FBS (PAA Laboratories, Linz, Austria). F11hTERT immortalized normal human fibroblasts, kindly provided by the laboratory of Dr Geza Safrany (National Research Institute for Radiobiology and Radiohygiene, Hungary), were grown in DMEM medium supplemented with 2 mM L-glutamine, 100 U ml−1penicillin, 100 μg ml−1streptomycin and 20% FBS. The cells were incubated in plastic flasks (Nunclon™, Nunc, Roskilde, Denmark) at 37 °C in a humidified atmosphere containing 5% CO2 and were subcultured every 2–3 days using Trypsin-EDTA solution. Experiments were carried out with cells up to 30 passages.
To determine the cytotoxicity of DCA, A431, MCF-7 and F11hTERT cells were seeded into 24-well plates (Costar, Corning) at densities of 2 × 103, 5 × 103, and 6 × 103cells per well, respectively, and were incubated for 48 h for proper attachment to the substratum. Afterwards, cells were washed once with phosphate buffered saline (PBS). One ml of medium with 10% FBS, containing DCA at a given concentration, was added to each well except controls. The cells were incubated for 48–96 h. Then, the cells were washed with PBS and incubated in the culture medium with MTT reagent (50 μl ml−1 of medium), without phenol red and FBS. After 2 h of incubation at 37 °C medium was removed and the cells were washed with PBS. Formazan crystals were reconstituted in 100% DMSO (400 μl per well). The plates were shaken for 20 min and 100 μl of supernatant was transferred into a 96-well plate and diluted with 100 μl of DMSO per well. The absorbance was measured at 540 nm on a Wallac Victor 2 plate reader (Perkin Elmer Corp., Norwalk, CT). Cell viability was estimated from the results of MTT assay and expressed as a percentage of viable treated cells relative to untreated control cells.
To determine whether the mitochondrial photodamage caused by ALA-PDT can be promoted by DCA, A431 and MCF-7 cells were seeded into 24-well plates (Costar, Corning) at densities of 5 × 103 and 7 × 103cells per well, respectively, and incubated for 48 h for proper attachment to the substratum. Afterwards, cells were washed twice with PBS; 0.5 ml of medium lacking FBS and phenol red but containing 1 mM of ALA was added to each well except controls. The cells were incubated for 3 h, washed twice with PBS and then 0.5 ml of medium without FBS and phenol red was added. Subsequently, the cells were exposed to light (400–460 nm, maximum at 420 nm, 10 mW cm−2) from four fluorescent tubes (Philips TLK 40 W/03, Eindhoven, The Netherlands). The light dose was 0.1 J cm−2 (MCF-7 cells) or 0.2 J cm−2 (A431 cells). Directly after light exposure fresh medium containing 10 mM of DCA, and supplemented with 10% FBS was added for another 48 h. Cell viability was determined with MTT assay as described above.
In all PDT experiments serum-free medium was used during ALA incubation and light exposure afterward to prevent a possible PpIX efflux.13,14Extracellular serum proteins bind endogenous PpIX molecules and increase the PpIX leakage from the cells.13–16
Each experiment was repeated at least two times with 4 parallels in each experimental group. The Statistica 8 software package (StatSoft Inc. 2007) was used for statistical analysis. The data were analyzed for normality with the Shapiro–Wilk's test. Levene's test was used to estimate the equality of variances between groups. t-Test for independent variables was used to compare means between different experimental groups. Values of p < 0.05 were considered statistically significant.
Cancer cell lines (A431, MCF-7) and immortalized normal human fibroblasts (F11hTERT) were grown in culture with increasing doses of DCA. Concentrations up to 1 mM were well tolerated by all cell lines and did not have any influence on viability (Fig. 1). Reduction in viability reached significance at 5 mM for A431 and 10 mM for MCF-7 cells (Fig. 2). As expected, DCA at 10 mM was safe for normal human cells in our in vitro model. Concentrations in the range of 20–100 mM significantly decreased cell viability between 2% and 27% depending on the cell line. Therefore, an approximate effective minimal dose of DCA for the cancer cells under the treatment period was determined to be between 5–10 mM. LD50 doses are calculated and presented in Table 1. Human squamous carcinoma cells (A431) were the most susceptible to DCA. The non-cancerous cells (F11hTERT) required the highest concentrations of DCA to affect viability.
| Cell line | DCA LD50/mM |
|---|---|
| A431 | 23 |
| MCF-7 | 30 |
| F11hTERT | 40 |
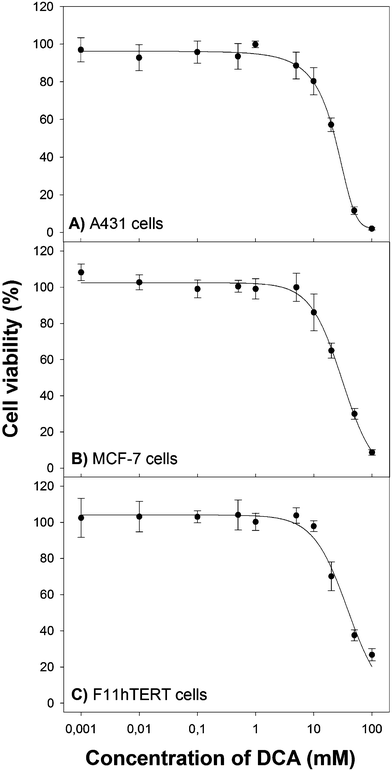 | ||
| Fig. 1 Cytotoxicity of DCA in (A) human squamous carcinoma cells A431, (B) human breast cancer cells MCF-7, and (C) human fibroblasts F11hTERT. The cells were incubated with various concentrations of DCA for 48 h in DMEM medium supplemented with 10% FBS. Cell viability was determined with the MTT assay. Error bars represent standard deviations (SD). Data points at various drug concentrations were fitted to a sigmoidal function using a three or four parameter logistic equation. | ||
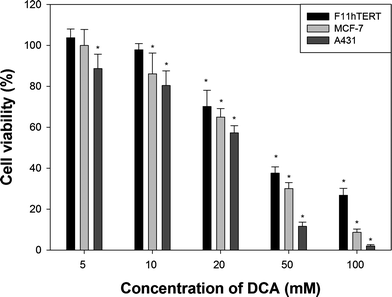 | ||
| Fig. 2 A comparison of cell viability of different cell lines after 48 h of treatment with selected concentrations of DCA. Error bars represent standard deviations (SD). * indicates statistically important differences (p < 0.05) when compared with untreated control. | ||
The outcome of ALA-PDT combined with DCA treatment is shown in Fig. 3. Cancer cells were incubated with 1 mM ALA for 3 h and then exposed to light. The light dose was adjusted to reduce cell viability up to 50–80% (0.1 J cm−2 for MCF-7 cells, 0.2 J cm−2 for A431 cells). Afterwards, the cells were cultured in medium containing 10 mM of DCA for 48 h. An additive effect of combined treatment was found. PDT alone reduce the viability of A431 cells to 83% at 0.2 J cm−2. Subsequent incubation with DCA decreased the viability to 60%, while DCA alone reduced cell survival to 81%. MCF-7 cells were considerably more susceptible to PDT (51% at 0.1 J cm−2), but more resistant to DCA (a decrease in viability to 89% was found). Combined treatment resulted in a viability of ∼38%. All differences in combined treatment were highly statistically significant when compared with PDT or DCA alone (p < 0.05). Light exposure alone had no influence on cell viability. PDT combined with 1 mM DCA treatment had no influence on overall outcome (data not shown). Extension of the incubation time with DCA up to 96 h slightly increased treatment effectiveness (data not shown).
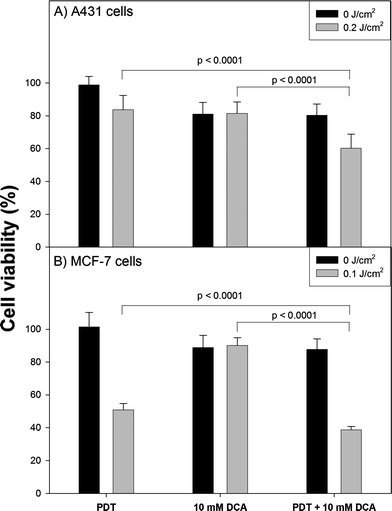 | ||
| Fig. 3 The outcome of PDT combined with metabolic-targeted therapy with DCA. A431 and MCF-7 cells were incubated with 1 mM ALA in serum-free medium for 3 h. Then the cells were exposed to light (400–460 nm, maximum at 420 nm, 10 mW cm−2). Directly after irradiation fresh medium containing 10 mM of DCA, supplemented with 10% FBS was added for 48 h. Cell viability was determined with MTT assay. Error bars represent standard deviations (SD). | ||
Since 2007, DCA has been focused on as a potential anticancer drugs, aiming at tumor cell metabolism. In preclinical studies, carried out on xenograft models, DCA showed significant anticancer effects in vitro and in vivo.8,17 There are several ongoing or completed clinical trials with DCA in the treatment of brain tumors, metastatic breast cancer, and lung cancer (Table 2). Recently, Dhar and Lippard18 described a novel drug resulting from the fusion of cisplatin and DCA, and called Mitaplatin. It displays a dual-killing mode that can only be effective in cancer cells. The platinum centre interacts with nuclear DNA, and the DCA released upon reduction attacks mitochondria.
| Targeted types of tumor | Phase of the study | Intervention | Study number |
|---|---|---|---|
| Recurrent and/or metastatic and pretreated breast and non-small cell lung cancer (NSCL) | II | Herceptin plus DCA (breast cancer); DCA (NSCL) | NCT01029925 |
| Malignant gliomas and glioblastome multiforme (GBM) | II | DCA | NCT00540176 |
| Recurrent and/or metastatic solid tumours | I | DCA | NCT00566410 |
| Recurrent malignant brain tumors | I | DCA | NCT01111097 |
| Newly diagnosed GBM | I | Radiotherapy plus Temozolomide plus DCA | NCT00703859 |
| Recurrent head and neck cancers | I | DCA | NCT01163487 |
In the present study we have shown for the first time that the outcome of PDT can be improved by DCA treatment. The minimal effective dose of DCA for A431 cells was 5 mM, and 10 mM for MCF-7 cells. Clinical trials on DCA toxicity in cancer patients are underway (NCT00566410, see Table 2). However, in vivo studies have shown that effective dose of DCA is in the range of 50–200 mg kg−1 day−1 with estimated plasma concentrations between 0.3–3 mM.8,17 This is similar to that in humans receiving 25 mg kg−1 day−1. Effective in vitro doses of DCA, not influencing viability of non-cancerous cells, vary from 0.5 to 10 mM.8,17,19,20 This is consistent with our results. DCA at the concentration of 10 mM reduced viability of both cancer cell lines but not normal human fibroblasts (F11hTERT).
Generally DCA is well tolerated. However, there was a high incidence of peripheral neuropathy in adults with MELAS (mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes) after administration of DCA at 25 mg kg−1 day−1 for 6 months, but peripheral neuropathy is a part of the MELAS syndrome.21 On the contrary, no peripheral neuropathy was observed in children with congenital acidosis after prolonged treatment with DCA at 25 mg kg−1 day−1.22
DCA was used after PDT, and an additive effect of the combined treatment was observed. DCA affects cancer cell metabolism, increases oxygen consumption, and, therefore makes the tumor more hypoxic. Increasing hypoxia may alleviate the outcome of therapies for which the presence of oxygen is required, like radiotherapy or PDT.10 Nevertheless, Cao et al.19 showed that DCA sensitizes prostate cancer cellsin vitro to radiation. However, cells grown in culture are generally in a constant atmosphere of about 18% oxygen23 which can freely diffuse into the cells. This is quite different from the situation in tumors. Several studies have revealed that DCA promotes proliferation of cancer cells under hypoxic conditions in vitro24,25 and in vivo.25 These results raise doubts about the use of DCA in cancer therapy. However, most in vivo studies have proved that DCA is an effective anticancer agent.8,17,26
We speculate that the outcome of PDTin vivo followed DCA treatment might be greater than in vitro. DCA should affect the cells which avoided direct (necrosis, apoptosis, autophagy) or indirect (immune responses, vascular effects) PDT cell killing mechanisms, thus reducing the risk of cancer recurrence and metastases. Moreover, DCA is already in clinical use, hence it can be easily combined with PDT treatment. Further studies are needed to determine mechanisms, optimal conditions and in vivo effectives of this novel therapeutic combination.
Abbreviations
| PpIX | Protoporphyrin IX |
| PDT | Photodynamic therapy |
| PS | Photosensitizer |
References
- M. Kwitniewski, A. Juzeniene, R. Glosnicka and J. Moan, Immunotherapy: a way to improve the therapeutic outcome of photodynamic therapy?, Photochem. Photobiol. Sci., 2008, 7, 1011–1017 RSC.
- M. Kwitniewski, D. Jankowski, K. Jaskiewicz, H. Dziadziuszko, A. Juzeniene, J. Moan, L. W. Ma, R. Peksa, D. Kunikowska, A. Graczyk, M. Kwasny, M. Kaliszewski and R. Glosnicka, Photodynamic therapy with 5-aminolevulinic acid and diamino acid derivatives of protoporphyrin IX reduces papillomas in mice without eliminating transformation into squamous cell carcinoma of the skin, Int. J. Cancer, 2009, 125, 1721–1727 CrossRef CAS.
- R. H. Xu, H. Pelicano, Y. Zhou, J. S. Carew, L. Feng, K. N. Bhalla, M. J. Keating and P. Huang, Inhibition of glycolysis in cancer cells: a novel strategy to overcome drug resistance associated with mitochondrial respiratory defect and hypoxia, Cancer Res., 2005, 65, 613–621 CAS.
- C. Ruckenstuhl, S. Buttner, D. Carmona-Gutierrez, T. Eisenberg, G. Kroemer, S. J. Sigrist, K. U. Frohlich and F. Madeo, The Warburg effect suppresses oxidative stress induced apoptosis in a yeast model for cancer, PLoS One, 2009, 4, e4592 CrossRef.
- E. D. Michelakis, L. Webster and J. R. Mackey, Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer, Br. J. Cancer, 2008, 99, 989–994 CrossRef CAS.
- P. W. Stacpoole, L. R. Gilbert, R. E. Neiberger, P. R. Carney, E. Valenstein, D. W. Theriaque and J. J. Shuster, Evaluation of long-term treatment of children with congenital lactic acidosis with dichloroacetate, Pediatrics, 2008, 121, e1223–8 CrossRef.
- E. Hassoun, C. Kariya and F. E. Williams, Dichloroacetate-induced developmental toxicity and production of reactive oxygen species in zebrafish embryos, J. Biochem. Mol. Toxicol., 2005, 19, 52–58 CrossRef CAS.
- S. Bonnet, S. L. Archer, J. Allalunis-Turner, A. Haromy, C. Beaulieu, R. Thompson, C. T. Lee, G. D. Lopaschuk, L. Puttagunta, S. Bonnet, G. Harry, K. Hashimoto, C. J. Porter, M. A. Andrade, B. Thebaud and E. D. Michelakis, A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth, Cancer Cell, 2007, 11, 37–51 CrossRef CAS.
- R. A. Cairns, I. Papandreou, P. D. Sutphin and N. C. Denko, Metabolic targeting of hypoxia and HIF1 in solid tumors can enhance cytotoxic chemotherapy, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 9445–9450 CrossRef CAS.
- N. C. Denko, Hypoxia, HIF1 and glucose metabolism in the solid tumour, Nat. Rev. Cancer, 2008, 8, 705–713 CrossRef CAS.
- D. Nowis, M. Makowski, T. Stoklosa, M. Legat, T. Issat and J. Golab, Direct tumor damage mechanisms of photodynamic therapy, Acta Biochim. Pol., 2005, 52, 339–352 CAS.
- A. P. Castano, T. N. Demidova and M. R. Hamblin, Mechanisms in photodynamic therapy: Part three - Photosensitizer pharmacokinetics, biodistribution, tumor localization and modes of tumor destruction, Photodiagn. Photodyn. Ther., 2005, 2, 91–106 CrossRef CAS.
- P. Steinbach, H. Weingandt, R. Baumgartner, M. Kriegmair, F. Hofstadter and R. Knuchel, Cellular fluorescence of the endogenous photosensitizer protoporphyrin IX following exposure to 5-aminolevulinic acid, Photochem. Photobiol., 2008, 62, 887–895 CrossRef.
- N. Schoenfeld, R. Mamet, Y. Nordenberg, M. Shafran, T. Babushkin and Z. Malik, Protoporphyrin biosynthesis in melanoma B16 cells stimulated by 5-aminolevulinic acid and chemical inducers: characterization of photodynamic inactivation, Int. J. Cancer, 2007, 56, 106–112 CrossRef.
- J. Hanania and Z. Malik, The effect of EDTA and serum on endogenous porphyrin accumulation and photodynamic sensitization of human K562 leukemic cells, Cancer Lett., 1992, 65, 127–131 CrossRef CAS.
- S. Iinuma, S. S. Farshi, B. Ortel and T. Hasan, A mechanistic study of cellular photodestruction with 5-aminolaevulinic acid-induced porphyrin, Br. J. Cancer, 1994, 70, 21–28 CAS.
- R. C. Sun, M. Fadia, J. E. Dahlstrom, C. R. Parish, P. G. Board and A. C. Blackburn, Reversal of the glycolytic phenotype by dichloroacetate inhibits metastatic breast cancer cell growth in vitro and in vivo, Breast Cancer Res. Treat., 2010, 120, 253–260 CrossRef CAS.
- S. Dhar and S. J. Lippard, Mitaplatin, a potent fusion of cisplatin and the orphan drug dichloroacetate, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 22199–22204 CrossRef CAS.
- W. Cao, S. Yacoub, K. T. Shiverick, K. Namiki, Y. Sakai, S. Porvasnik, C. Urbanek and C. J. Rosser, Dichloroacetate (DCA) sensitizes both wild-type and over expressing Bcl-2 prostate cancer cells in vitro to radiation, Prostate, 2008, 68, 1223–1231 CrossRef CAS.
- J. Y. Wong, G. S. Huggins, M. Debidda, N. C. Munshi and I. De Vivo, Dichloroacetate induces apoptosis in endometrial cancer cells, Gynecol. Oncol., 2008, 109, 394–402 CrossRef CAS.
- P. Kaufmann, K. Engelstad, Y. Wei, S. Jhung, M. C. Sano, D. C. Shungu, W. S. Millar, X. Hong, C. L. Gooch, X. Mao, J. M. Pascual, M. Hirano, P. W. Stacpoole, S. DiMauro and D. C. De Vivo, Dichloroacetate causes toxic neuropathy in MELAS: a randomized, controlled clinical trial, Neurology, 2006, 66, 324–330 CrossRef CAS.
- P. W. Stacpoole, D. S. Kerr, C. Barnes, S. T. Bunch, P. R. Carney, E. M. Fennell, N. M. Felitsyn, R. L. Gilmore, M. Greer, G. N. Henderson, A. D. Hutson, R. E. Neiberger, R. G. O'Brien, L. A. Perkins, R. G. Quisling, A. L. Shroads, J. J. Shuster, J. H. Silverstein, D. W. Theriaque and E. Valenstein, Controlled clinical trial of dichloroacetate for treatment of congenital lactic acidosis in children, Pediatrics, 2006, 117, 1519–1531 CrossRef.
- D. Newby, L. Marks and F. Lyall, Dissolved oxygen concentration in culture medium: assumptions and pitfalls, Placenta, 2005, 26, 353–357 CrossRef CAS.
- K. M. Anderson, J. Jajeh, P. Guinan and M. Rubenstein, In vitro effects of dichloroacetate and CO2 on hypoxic HeLa cells, Anticancer Res., 2009, 29, 4579–4588 CAS.
- S. Shahrzad, K. Lacombe, U. Adamcic, K. Minhas and B. L. Coomber, Sodium dichloroacetate (DCA) reduces apoptosis in colorectal tumor hypoxia, Cancer Lett., 2010 Search PubMed.
- E. D. Michelakis, G. Sutendra, P. Dromparis, L. Webster, A. Haromy, E. Niven, C. Maguire, T. L. Gammer, J. R. Mackey, D. Fulton, B. Abdulkarim, M. S. McMurtry and K. C. Petruk, Metabolic modulation of glioblastoma with dichloroacetate, Sci. Transl. Med., 2010, 2, 31ra34 Search PubMed.
| This journal is © The Royal Society of Chemistry and Owner Societies 2011 |