Synthesis of enantioenriched γ-quaternary cycloheptenones using a combined allylic alkylation/Stork–Danheiser approach: preparation of mono-, bi-, and tricyclic systems†
Nathan B.
Bennett
,
Allen Y.
Hong
,
Andrew M.
Harned
and
Brian M.
Stoltz
*
The Warren and Katharine Schlinger Laboratory for Chemistry and Chemical Engineering, California Institute of Technology, Division of Chemistry and Chemical Engineering, Mail Code 101-20, Pasadena, CA 91125, USA. E-mail: stoltz@caltech.edu; Fax: +1-626-395-8436; Tel: +1-626-395-6064
First published on 1st September 2011
Abstract
A general method for the synthesis of β-substituted and unsubstituted cycloheptenones bearing enantioenriched all-carbon γ-quaternary stereocenters is reported. Hydride or organometallic addition to a seven-membered ring vinylogous ester followed by finely tuned quenching parameters achieves elimination to the corresponding cycloheptenone. The resulting enones are elaborated to bi- and tricyclic compounds with potential for the preparation of non-natural analogs and whose structures are embedded in a number of cycloheptanoid natural products.
Introduction
Cycloheptanes bearing all-carbon quaternary stereocenters are incorporated into the polycyclic cores of many natural products including the daphnicyclidins (2),1a guanacastepenes (3),1b–d cyathins (4),1e–i dhilirolide D (5),1jtricholomalide B (6),1k–l miniolutelide A (7),1m and berkeleydione (8)1m–o (Fig. 1A). Due to the biological relevance and structural complexity of these compounds, we sought to develop a stereoselective approach to this quaternary carbon-containing cycloheptane motif. To this end, we envisioned cycloheptenone 1 as a promising synthetic intermediate. Additionally, we viewed enone 1 as an attractive scaffold for annulation strategies toward bi- and tricyclic structures potentially valuable in total synthesis and the preparation of non-natural analogs. Particularly attractive to us was the homologous structural relationship of the desired [7 – n] bicyclic scaffold to the classic [6 – 5] and [6 – 6] frameworks used in hundreds of approaches to natural products (Fig. 1B). Herein we describe a general enantioselective route to cycloheptenone 1 that allows for facile elaboration to bi- and tricyclic products. | ||
| Fig. 1 Potential applications of cycloheptenone 1. | ||
Retrosynthetically, we planned to access cycloheptenone 1 using a Stork–Danheiser type transposition of vinylogous ester 9 (Scheme 1A). In this approach, the quaternary stereocenter of vinylogous ester 9 would be installed by employing our palladium-catalyzed asymmetric allylic alkylation methodology.2,3 Toward this end, acylation and alkylation of vinylogous ester 10 generates racemic β-ketoester 11, which under our standard decarboxylative alkylation conditions is converted to vinylogous ester 9 (Scheme 1B).2d
 | ||
| Scheme 1 Retrosynthesis for cycloheptenone 1 and route to vinylogous ester 9. | ||
As previously reported,2d initial efforts toward cycloheptenone 1 employing standard transposition conditions4 were unsuccessful and led to the discovery of the unusual reactivity of vinylogous ester 9. In contrast to the six-membered ring analog (12), both reduction and organometallic addition to vinylogous ester 9 followed by strong acidic work-up favor formation of the corresponding β-hydroxyketones (14a and 14b) instead of the cycloheptenones (1a and 1b) (Scheme 2A and B). Attempts to convert the β-hydroxyketones to enones resulted in an unexpected retro-aldol/aldol ring contraction that our lab has examined extensively (Scheme 2C).2d This unexpected reactivity prompted further investigation of the reaction sequence to develop new conditions for the efficient preparation of cycloheptenones.
 | ||
| Scheme 2 Previous investigation into reactivity of vinylogous ester 9 and β-hydroxyketone 14. | ||
Results and discussion
After some investigation, alternative reaction conditions enabled access to the elusive β-unsubstituted and substituted cycloheptenones. Gratifyingly, Luche reduction of vinylogous ester 9 followed by strong acidic work-up preferentially generated unsubstituted enone 1a (Scheme 3).5 Additionally, quenching the Grignard reaction of vinylogous ester 9 with a sodium phosphate buffer and treating the resulting crude oil with dilute acid in acetonitrile afforded substituted enone 1b. Analysis of the initial crude material suggests that hydroxy enol ether 16 is formed after the sodium phosphate buffer quench.6 Subsequent acid treatment likely protonates alcohol 16, which leads to dehydration via generation of the resonance stabilized tertiary carbocation and eventual collapse to enone 1b. Overall, these modified conditions provide divergent routes to γ-quaternary cycloheptenones and acylcyclopentenes in conjunction with our preceding work.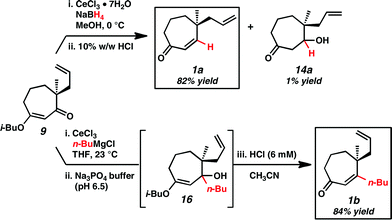 | ||
| Scheme 3 Reduction and organometallic addition conditions favoring cycloheptenone formation. | ||
Based on these initial results, we examined the scope of β-substituted enones available from nucleophilic attack on vinylogous ester 9. The buffer and dilute acid conditions described above (Table 1, method A) accommodate β-substituent groups initiating in sp3 hybridization, producing allyl, homoallyl, and pentenyl substituted enones in moderate to excellent yield (entries 1–5). Attempts to apply this quenching sequence to reactions involving sp and sp2 hybridized carbon nucleophiles resulted in complex reaction mixtures. Selectivity for the cycloheptenone was restored by quenching such reactions with a concentrated strong acid (i.e., hydrochloric or sulfuric acid) and heating the resulting solutions at elevated temperature (Table 1, methods B and C). These conditions initially produce a mixture of β-hydroxyketone and enone that converges to the desired product over time.7 In this manner, the synthesis of vinyl, alkynyl, aryl, and heteroaryl substituted enones is accomplished (entries 6–10). Of particular note is entry 8, in which an ortho-substituted aryl Grignard reagent can be incorporated to generate enone 1j.8 In general, application of the appropriate work-up conditions allows access to a variety of β-substituted γ-quaternary cycloheptenones.
|
|
||||
|---|---|---|---|---|
| Entry | R | Work-upb | Product (11) | Yield (%)c |
a Conditions: vinylogous ester 9 (1.0 equiv), CeCl3 (2.5 equiv), RMgX or RLi (3.0 equiv) in THF, 23 °C then work-up by methods A, B, or C.
b Method A: a) pH 6.5 Na3PO4 buffer b) 6 mM HCl, CH3CN; Method B: 10% w/w aq HCl, 60 °C; Method C: 2 M H2SO4, 60 °C.
c Yield of isolated product.
d See Supporting Information for slightly different reaction parameters.
e Product is 1.9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of atropisomers. :1 mixture of atropisomers.
|
||||
| 1 |

|
A | 1c1c | 73 |
| 2 |
|
A | 1d1d | 93 |
| 3 |

|
A | 1e1e | 90 |
| 4 |

|
A | 1f1f | 82 |
| 5 |

|
A | 1g1g | 92 |
| 6d |

|
B | 1h1h | 84 |
| 7 |

|
C | 1i1i | 97 |
| 8e |

|
C | 1j1j | 66 |
| 9 |
|
B | 1k1k | 72 |
| 10 |

|
B | 1l1l | 84 |

With various cycloheptenones in hand, we sought to elaborate these compounds to bi- and tricyclic structures (Table 2). We first examined olefin metathesis reactions between the β-substituent and quaternary center allyl fragment to generate a number of [7 – 5], [7 – 6], [7 – 7], and [7 – 8] fused ring systems. Substrates possessing two terminal olefins lead to bicyclic products with high efficiency (entries 1, 3, 5, and 8). This process also accommodates the production of trisubstituted olefin products (i.e., 17b, 17d, and 17f) through ring-forming enyne metathesis (entry 2) or ring closing metathesis (entries 4 and 6). In addition, cycloheptenone 1j is converted to the [7 – 7 – 6] tricyclic enone (17g) under the reaction conditions (entry 7). The ketone transposition/ring closing metathesis sequence is also amenable to trans-propenyl analog 18,9 producing the [7 – 6] system (17i) with the alkene adjacent to the quaternary center (Scheme 4A).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Substrate (11) | R1 | Product (1717) | Yield (%)b | ||
| a Conditions: cycloheptenone 1 (1.0 equiv) and Grubbs–Hoveyda 2nd generation catalyst (5.0 mol%) in benzene, 50 °C. b Yield of isolated product. c See Supporting Information for alternative reaction parameters. d 1,4-benzoquinone (10 mol%) added. e Performed in toluene. | ||||||
| 1c | 1h1h |

|

|
17a17a | R 2 = H | 93 |
| 2 | 1i1i |

|
17b17b |
|
99 | |
| 3d | 1c1c |

|
|
17c17c | R 2 = H | 91 |
| 4d | 1d1d |

|
17d17d | R 2 = Me | 90 | |
| 5 | 1e1e |

|

|
17e17e | R 2 = H | 90 |
| 6 | 1f1f |

|
17f17f | R 2 = Me | 98 | |
| 7e | 1j1j |
|

|
96 | ||
| 8 | 1g1g |

|

|
99 | ||

 | ||
| Scheme 4 Synthetic applications. | ||
Having produced two [7 – 6] structures with variable olefin positions, we next investigated conditions to generate the conjugated dienone system. Interestingly, treatment of skipped diene 17c with base at ambient temperature migrated both olefins into the six-membered ring, producing diene 17j (Scheme 4B). Alternatively, the alkenes can be migrated into conjugation with the carbonyl by microwave irradiation, affording diene 17k (Scheme 4B).
Lastly, we envisioned enone 1i as an ideal substrate for a Pauson–Khand reaction given the proximal enyne functionality. Treatment of 1i with dicobalt octacarbonyl employing dimethylsulfoxide as an activating agent10 produced the [7 – 5 – 5] tricycle in excellent yield with a 3:1 diastereomeric ratio of 19a:19b (Scheme 4C).
Conclusions
In summary, we have developed a method to access β-functionalized cycloheptenones (1) possessing a γ-quaternary stereocenter through a sequence involving asymmetric alkylation followed by addition of an organometallic reagent and acid-mediated ketone transposition. Subsequent manipulation of the newly incorporated β-substituents provides a number of bi- and tricyclic compounds with potential for the preparation of non-natural analogs and whose structure is present in cycloheptanoid natural products. Further efforts toward the total synthesis of such targets will be reported in due course.Acknowledgements
This publication is based on work supported by Award No. KUS-11-006-02, made by King Abdullah University of Science and Technology (KAUST). The authors wish to thank NIH-NIGMS (R01GM080269-01), Amgen, Abbott, Boehringer Ingelheim, and Caltech for financial support. AMH thanks the NIH for a postdoctoral fellowship. Materia, Inc. is gratefully acknowledged for the donation of catalysts. Michael Krout, Thomas Jensen, Christopher Henry, Scott Virgil, and Sarah Reisman are acknowledged for helpful discussions. David VanderVelde is acknowledged for critical NMR support.Notes and references
- (a) J. Kobayashi, Y. Inaba, M. Shiro, N. Yoshida and H. Morita, J. Am. Chem. Soc., 2001, 123, 11402 CrossRef CAS; (b) S. F. Brady, M. P. Singh, J. E. Janso and J. Clardy, J. Am. Chem. Soc., 2000, 122, 2116 CrossRef CAS; (c) M. R. Singh, J. E. Janso, S. W. Luckman, S. F. Brady, J. Clardy, M. Greenstein and W. M. Maiese, J. Antibiot., 2000, 53, 256 CAS; (d) S. F. Brady, S. M. Bondi and J. Clardy, J. Am. Chem. Soc., 2001, 123, 9900 CrossRef CAS; (e) A. D. Allbutt, W. A. Ayer, H. J. Brodie, B. N. Johri and H. Taube, Can. J. Microbiol., 1971, 17, 1401 CrossRef CAS; (f) W. A. Ayer and H. Taube, Tetrahedron Lett., 1972, 13, 1917 CrossRef; (g) W. A. Ayer and H. Taube, Can. J. Chem., 1973, 51, 3842 CrossRef CAS; (h) W. A. Ayer, T. T. Nakashima and D. E. Ward, Can. J. Chem., 1978, 56, 2197 CrossRef CAS; (i) H.-J. Hecht, G. Höfle, W. Steglich, T. Anke and F. Oberwinkler, J. Chem. Soc., Chem. Commun., 1978, 665 RSC; (j) E. D. de Silva, D. E. Williams, D. R. Jayanetti, R. M. Centko, B. O. Patrick, R. L. C. Wijesundera and R. Andersen, Org. Lett., 2011, 13, 1174 CrossRef; (k) S. Tsukamoto, A. D. Macabalang, K. Nakatani, Y. Obara, N. Nakahata and T. Ohta, J. Nat. Prod., 2003, 66, 1578 CrossRef CAS; (l) Z. Wang, S.-J. Min and S. J. Danishefsky, J. Am. Chem. Soc., 2009, 131, 10848 CrossRef CAS; (m) M. Iida, T. Ooi, K. Kito, S. Yoshida, K. Kanoh, Y. Shizuri and T. Kusumi, Org. Lett., 2008, 10, 845 CrossRef CAS; (n) D. B. Stierle, A. A. Stierle, J. D. Hobbs, J. Stokken and J. Clardy, Org. Lett., 2004, 6, 1049 CrossRef CAS; (o) D. B. Stierle, A. A. Stierle and B. Patacini, J. Nat. Prod., 2007, 70, 1820 CrossRef CAS.
- (a) D. C. Behenna and B. M. Stoltz, J. Am. Chem. Soc., 2004, 126, 15044 CrossRef CAS; (b) J. T. Mohr, D. C. Behenna, A. M. Harned and B. M. Stoltz, Angew. Chem., Int. Ed., 2005, 44, 6924 CrossRef CAS; (c) J. T. Mohr and B. M. Stoltz, Chem.–Asian J., 2007, 2, 1476 CrossRef CAS; (d) A. Y. Hong, M. R. Krout, T. Jensen, N. B. Bennett, A. M. Harned and B. M. Stoltz, Angew. Chem., Int. Ed., 2011, 50, 2756 CrossRef CAS; (e) D. E. White, I. C. Stewart, R. H. Grubbs and B. M. Stoltz, J. Am. Chem. Soc., 2008, 130, 810 CrossRef CAS; (f) J. A. Enquist Jr. and B. M. Stoltz, Nature, 2008, 453, 1228 CrossRef; (g) S. R. Levine, M. R. Krout and B. M. Stoltz, Org. Lett., 2009, 11, 289 CrossRef CAS; (h) K. V. Petrova, J. T. Mohr and B. M. Stoltz, Org. Lett., 2009, 11, 293 CrossRef CAS.
- Concurrent with our efforts, palladium-catalyzed asymmetric allylic alkylations to form enantioenriched α-quaternary vinylogous esters and thioesters have also been performed by Trost. See: (a) B. M. Trost, C. Pissot-Soldermann, I. Chen and G. M. Schroeder, J. Am. Chem. Soc., 2004, 126, 4480 CrossRef CAS; (b) B. M. Trost and G. M. Schroeder, Chem.–Eur. J., 2005, 11, 174 CrossRef; (c) B. M. Trost, C. Pissot-Soldermann and I. Chen, Chem.–Eur. J., 2005, 11, 951 CrossRef CAS; (d) B. M. Trost, R. N. Bream and J. Xu, Angew. Chem., Int. Ed., 2006, 45, 3109 CrossRef CAS.
- G. Stork and R. L. Danheiser, J. Org. Chem., 1973, 38, 1775 CrossRef CAS.
- Methanol likely plays a large role in selectivity inasmuch as exchanging the solvent to diethyl ether after Luche reduction and before acid treatment leads to the β-hydroxyketone preferentially. See ref. 2d.
- Determined by 1H NMR analysis of crude oil. Attempts to purify the crude material under several chromatography conditions resulted in decomposition to cycloheptenone 1b and β-hydroxyketone 14b.
- Disappearance of β-hydroxyketone can be monitored by TLC analysis.
-
Cycloheptenone
1j is formed as a 1.9:1 mixture of atropisomers. Broadening of the isomeric peaks was observed in a variable temperature 1H NMR study. See Supporting Information.
- trans-Propenyl analog 18 is prepared by palladium-catalyzed isomerization of terminal olefin 9. See ref. 2d for details.
- Y. K. Chung, B. Y. Lee, N. Jeong, M. Hudecek and P. L. Pauson, Organometallics, 1993, 12, 220 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data, copies of NMR and IR spectra for compounds synthesized in this study. See DOI: 10.1039/c1ob06189e |
| This journal is © The Royal Society of Chemistry 2012 |