Dynamic combinatorial libraries for the recognition of heavy metal ions†
Jörg M.
Klein
a,
Vittorio
Saggiomo‡
a,
Lisa
Reck
b,
Ulrich
Lüning
*b and
Jeremy K. M.
Sanders
*a
aUniversity Chemical Laboratory, University of Cambridge, Lensfield Road, Cambridge, UK CB2 1EW. E-mail: jkms@cam.ac.uk; Fax: +44 (0)1223 336 17
bOtto-Diels-Institut für Organische Chemie, Olshausenstr. 40, D-24098, Kiel, Germany. E-mail: luening@oc.uni-kiel.de; Fax: +49-431-880-1558; Tel: +49-431-880-2450
First published on 25th October 2011
Abstract
We present the use of hydrazone dynamic combinatorial libraries (DCLs) to identify macrocyclic receptors that are selective for alkaline earth metal ions over alkali metal ions. In particular, the toxic heavy metal ions Sr2+ and Ba2+ induce characteristic changes in the DCLs. Four macrocycles were isolated and characterised by LCMS, HRMS, NMR and X-ray crystallography; binding studies by UV-Vis spectroscopy confirm the selectivity observed in the DCLs.
1 Introduction
Heavy metal ions are part of everyday life and much research has focussed on the extraction of heavy metal ions from organic or aqueous effluents.1–7 In particular, radioactive isotopes such as 90Sr and 137Cs have gained renewed attention following recent accidents in the nuclear power plants in Japan. As recently pointed out by Chen et al., new organic ligands are needed to further optimise the extraction and make it a greener process.8 Non-covalent interactions play an important role in the recognition between the metal and the organic ligand and have been found to be a way of tuning the extractant strength.9 We now present the use of dynamic combinatorial chemistry (DCC) to discover new organic ligands that are selective for heavy metal ions and could contribute to more efficient extraction processes; some of these results have previously been presented in preliminary form.10Dynamic combinatorial chemistry is concerned with mixtures of building blocks under thermodynamic control.11,12 The building blocks can assemble to give different combinations (in this case macrocycles). The assembly process is reversible thus making the mixture dynamic. These adaptable mixtures are called dynamic combinatorial libraries (DCLs) and adapt to external stimuli (e.g. temperature, pressure, the presence of a template etc.) by changing the product distribution to minimise the total energy of the system. The increase in concentration of a particular species is referred to as amplification. Amplification reflects stabilisation of a particular species and has guided the discovery of new receptors.13–27
The reversible interactions used in this study are hydrazone exchange23–27 and metal–ligand interactions.28 Recently, we exploited the reversibility of the hydrazone linkage in ligands containing a 2,6-bis-hydrazonopyridine core (here referred to as N3O2 ligands)29–35 to study their behaviour in dynamic combinatorial libraries (DCLs).10Dialdehyde 1 and dihydrazide 2a (Fig. 1) were combined to generate two different macrocycles. We developed a protocol for the easy isolation of the first macrocyclic hydrazone N3O2 ligand and presented its characterisation. Binding studies showed a striking selectivity of this host for Ba2+ over Ca2+ and alkali metal ions, and proved the potential of macrocyclic N3O2 ligands.10
 | ||
| Fig. 1 Schematic representation of the experiment. A mixture of macrocycles is formed by combining a dialdehyde with a dihydrazide. Addition of a metal ion leads to the selection of the macrocycle that binds the metal ion better. | ||
Here we extend the family of macrocyclic N3O2 ligands by introducing a new dihydrazide building block 2b (with a diethyleneglycol linker) and study DCLs that were templated with alkali and alkaline earth metal ions (Fig. 1). The heavy metal ions Sr2+ and Ba2+ induced characteristic changes in the product distribution of the DCLs that were not observed when other metal ions were added. After the isolation and characterisation of four different N3O2 macrocycles, binding studies of the larger 2 + 2 macrocycles by HRMS, NMR and UV-Vis confirmed the selectivity of the macrocycles for Sr2+ and Ba2+.
2 Templating of DCLs with metal ions
The building blocks were synthesised according to published procedures10,36 or as described in the ESI.† Dissolution of dialdehyde 1 and dihydrazide (2a or 2b) in mixtures of CHCl3/MeOH/F3CCO2H generated two different sets of DCLs (Fig. 2 and ESI†). As shown by LCMS analysis, both sets of untemplated DCLs consisted of almost exclusively 1 + 1 macrocycle (3a and 3b); therefore these smallest macrocycles, which are inevitably favoured on entropic grounds, represent the thermodynamically most stable species (Fig. 2 and ESI†). Addition of alkali metal ions had essentially no effect on the product distribution whereas addition of alkaline earth metal ions stabilised larger macrocycles and increased their abundance in the DCLs. Interestingly, the small differences in the linker unit of 2a and 2b led to distinctly different responses of the DCLs to templating with Ca2+, Sr2+ and Ba2+. The DCLs with the ethylene glycol linker (Fig. 2) showed amplification of 2 + 2 (4b), 3 + 3 (5b) and 4 + 4 (6b) macrocycles upon addition of Ba2+ and Sr2+ while the DCLs with the pyridyl linker showed only amplification of the 2 + 2 macrocycle 4a (ESI†).10 These results suggested that the 2 + 2 macrocycle with the pyridyl linker and the 2 + 2, 3 + 3 and 4 + 4 macrocycles with the ethylene glycol linker are good hosts for the doubly charged guests Ca2+, Sr2+ and Ba2+ while showing weak or no binding to singly charged guests (alkali metal ions). Thermodynamic control was demonstrated by reaching the same library distribution from two different starting points. When the 2 + 2 macrocycles (isolation is described below) were dissolved under the same conditions as the building blocks, an identical product distribution was reached (ESI†). The following section will present the isolation and characterisation of the two 1 + 1 macrocycles 3a and 3b and the two 2 + 2 macrocycles 4a and 4b. | ||
| Fig. 2 LCMS analysis of DCLs generated from building blocks 1 and 2b in the absence and presence of different metal ions. Cartoons as explained in Fig. 1. Method A (ESI†). | ||
3 Isolation and characterisation of macrocycles
3.1 1 + 1 Macrocycles
The isolation and characterisation of the 1 + 1 macrocycle with the pyridyl linker 3a was very similar to that of the 1 + 1 macrocycle with the ethylene glycol linker 3b and only the latter is described here. The information for 3a can be found in the ESI.† The untemplated DCL of 1 and 2b formed almost exclusively 1 + 1 macrocycle 3b as the thermodynamically most stable species and the 1 + 1 macrocycles were isolated in a simple three-step procedure: addition of base (to halt hydrazone exchange) followed by removal of solvent and finally removal of salts by washing with water. LCMS analysis confirmed that the isolated material was indeed the 1 + 1 macrocycle 3b and that the purity of the obtained sample was high (ESI,† 99%).The NMR spectrum of 3b showed that this macrocycle adopts an asymmetric conformation. NOESY cross-peaks (Fig. 3) for only one of the two imine peaks with the meta-pyridine protons suggested that the asymmetry arises through different orientations of the hydrazone moiety on the two sides of the MeO-pyridine ring.
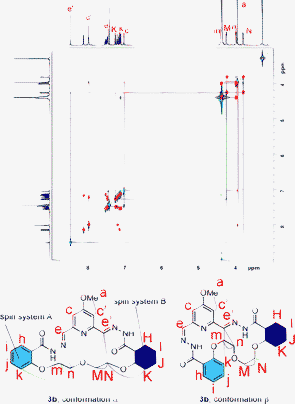 | ||
Fig. 3 Overlaid COSY (red cross peaks) and NOESY (blue peaks) spectra of 3b in CDCl3/MeOD (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 500 MHz, 297 K, mixing time = 800 ms). Cross-peaks are indicated by corresponding colours in the spectrum and the suggested structures below. :1, 500 MHz, 297 K, mixing time = 800 ms). Cross-peaks are indicated by corresponding colours in the spectrum and the suggested structures below. | ||
The resonances of the two spin systems A and B could not be assigned unambiguously by COSY/NOESY and two possible conformations are shown in Fig. 3.
The solid state structure (Fig. 4) of the macrocycle with the ethylene glycol linker 3b showed an asymmetric structure, the two substituents on the pyridine ring adopting different conformations. The structure obtained from X-ray crystallography is very similar to the NMR-derived conformation β in Fig. 3, suggesting that the macrocycle adopts similar conformations in solution and solid state.
 | ||
| Fig. 4 Solid state structure of the 1 + 1 macrocycle 3b.† a) top view; b) side view; c) different conformations of hydrazones moieties in the same macrocycle and important H-bonds (dotted lines). | ||
3.2 2 + 2 Macrocycles
Varying building block concentrations in these DCLs (Fig. 5) showed that, if the concentration of the building blocks were increased, the 2 + 2 macrocycle 4b precipitated, creating a useful kinetic trap. The precipitation of 4b shifts the equilibrium towards its formation, consuming all the building blocks. The 2 + 2 macrocycles were easily isolated by filtration. The group of Nitschke used this principle (i.e. isolation of a compound by exploiting a kinetic trap) to convert a dynamic library of six diastereomeric pairs of enantiomers into a single pair of enantiomers.37LCMS analysis of the precipitated macrocycles showed that the purity of the obtained samples was satisfactory (95% for 4b, ESI†). | ||
| Fig. 5 HPLC analysis of DCLs generated from 1 and 2b at different building block concentrations. At low building block concentrations the DCLs are homogeneous and the 1 + 1 macrocycle 3b is the only detected product. At concentrations above 5 mM the larger macrocycle 4b precipitates, forming a kinetic trap. Analysis of the mixtures (precipitate and solution) showed that 4b has become more abundant than 3b. | ||
The NMR spectra of the larger 2 + 2 macrocycles 4a and 4b showed a time-averaged symmetrical structure exhibiting only half as many peaks as their smaller 1 + 1 analogues 3a and 3b. This might reflect the symmetrical orientation of the hydrazone substituents on the pyridine core or the flexibility of the macrocycles which exist as mixtures of rapidly interconverting conformations so that only a time average of these conformations is observed. The cross peak between protons c and e that was used to gain information about the conformation of the hydrazones in Fig. 3 was analysed similarly in the NOESY spectra of 4a and 4b (Fig. 6). However, the information from these two NOESY spectra cannot be compared easily because the spectra were recorded in different solvent mixtures to maximise the solubility of the macrocycles in the NMR solvent. A cross-peak between c and e was present in the NOESY spectrum of 4b (Fig. 6b), but not in the NOESY spectrum of 4a (Fig. 6a) suggesting that the conformation of the hydrazones depends on the linker unit. Both NOESY spectra showed cross-peaks between aromatic rings suggesting that aromatic interactions influence the folding of the macrocycles in solution.
 | ||
| Fig. 6 Partial NOESY spectra of 4a and 4b in CDCl3/MeOD (1:1 and 7:3 respectively, 500 MHz, 297 K, mixing time = 800 ms). Cross-peaks are indicated by corresponding colours in the spectrum and the suggested structures below. | ||
The solid state structure of the macrocycle with the ethylene glycol linker 4b (Fig. 7) shows clearly different orientations of hydrazones in the same macrocycle. The only structural information concerning unbound N3O2 ligands in the literature comes from two crystal structures.38,39 In these (both are linear N3O2 ligands) the hydrazones point away from each other, requiring large conformational changes upon binding of metal ion guests.29–35 The differences between the published linear ligand and the macrocycles (3a, 3b, 4a, 4b) in this study can be interpreted as a result of ring strain imposed on the N3O2 binding motif by the linker unit. To bind the guest strongly the macrocycles need the right balance of preorganisation and flexibility. All four macrocycles (3a, 3b, 4a, 4b) have a partially preorganised binding pocket but the 2 + 2 macrocycles 4a and 4b are larger and more flexible and are amplified at the expense of the 1 + 1 macrocycles. Consequently the right linker unit could increase the ability of N3O2 ligands to bind metal ion guests by partly preorganising the binding pocket and still maintaining some flexibility. The intramolecular hydrogen bonds that are observed in the crystal structure of 4b are expected to be weakened in solution, but they might still play an important role in preorganising 4b for binding.
 | ||
| Fig. 7 Solid state structure of 4b.† a) top view (perpendicular to the plane of py); b) side view (parallel to the plane of py); c) and d) different conformations of hydrazones moieties in the same macrocycle and important H-bonds (dotted lines). | ||
4 Binding to alkaline earth metal ions
The binding of the 2 + 2 macrocycles 4a and 4b to alkaline earth metal ions was studied by NMR, HRMS and UV-Vis. To ensure stability of the isolated macrocycles, the binding studies had to be performed in a different solvent system than the DCLs (i.e. without acid). Since protonated species are significantly less stabilised in organic solvents compared to water we do not expect the protonation states of the macrocycles to be very different. Switching from 2-methoxyethanol/water (80/20) to ethanol decreases the pKa values of carboxylic acid by ≈4.40 Since we work in even less polar solvent mixtures the stabilisation of charged species is not expected to be significant.4.1 UV-Vis
To quantify the binding affinity between the host macrocycles (4a and 4b) and metal ion guests we performed UV-Vis titrations (ESI† and Fig. 8) and fitted the obtained binding isotherms to a published model.41 The macrocycle with the pyridyl linker 4a bound weakly to Mg2+ and Ca2+ (ESI,†Ka ≈ 103M−1) and strongly to Sr2+ and Ba2+ (ESI†, Ka ≈ 105–106M−1) with one of the best Ba2+/Ca2+ selectivities reported to date.10Macrocycle 4b bound Mg2+ with an affinity of Ka ≈ 105M−1. The binding constants between Ca2+, Sr2+, Ba2+ and 4b were too large to be determined accurately with this method and Ka ≈ 106M−1 is given as a lower limit (ESI,†Fig. 8). Besides proving the strong affinity of the macrocyclic hosts 4a and 4b for selected alkaline earth metal ions the binding isotherms strongly suggested a 1:1 binding stoichiometry. Surprisingly, the selectivity of 4a for Ba2+ over Ca2+ is not found in its closely related diethyleneglycol analogue 4b. It might seem surprising that 4b shows similar affinities for Ca2+, Sr2+ and Ba2+ while the amplification patterns in the DCL are different. The two results are consistent if one considers that the product distribution in the DCL is not only determined by the interaction of the 2 + 2 macrocycle 4b with the template but by all interactions between the library members.42 The small Ca2+ ion binds only to the 2 + 2 macrocycle 4b while the larger Sr2+ and Ba2+ ions also bind to 5b and 6b in addition to 4b. Therefore, Sr2+ and Ba2+ amplify 4b, 5b and 6b whereas Ca2+ amplifies only 4b. Furthermore, the metals that amplified 4b in the DCLs also showed affinity in the UV-Vis study, showing that good receptors can be discovered by DCC when keeping in mind that DCLs are complex systems.43–46
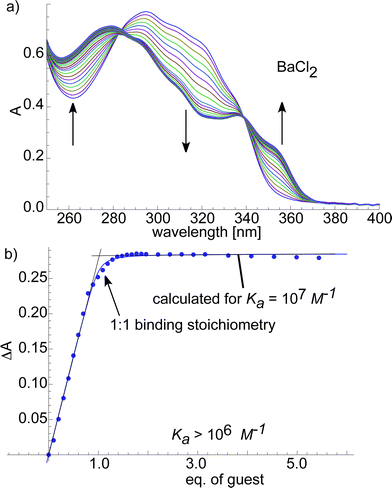 | ||
| Fig. 8
UV-Vis
titrations of ethylene glycol-based 4b with BaCl2 in CHCl3/MeOH (7:3). a) UV-Vis spectra of 4b with different amounts of BaCl2. Increasing and decreasing bands are indicated by arrows. b) Binding isotherm resulting from a plot of the change in absorption ΔA at 320 nm against the guest concentration. Experimental data represented as dots and calculated binding isotherm represented as a solid line. | ||
Comparing 4a with 4b shows that small differences in the linker units can lead to unpredictably different host–guest chemistries. These differences can be observed in the DCLs as well as when the isolated compounds are studied.
4.2 HRMS and NMR
After addition of 1.0 eq. of metal ion salt (MgCl2, CaCl2, SrCl2 and BaCl2) to 4a and 4b, the Ba2+ and Sr2+ complexes (1:1) were observed by HRMS as singly (loss of one proton) and doubly charged molecular ion peaks. The complexes of Mg2+ and Ca2+ with 4a and 4b were not observed by ESI-HRMS.
Well-resolved NMR spectra were obtained when SrCl2 or BaCl2 were added to solutions of 4a (ESI†) or 4b (Fig. 9). The largest shifts upon addition of the metal salt were observed for protons c, e and h confirming the involvement of the N3O2 binding motif. A possible explanation for the observed shifts is that c and e are shifted through inductive effects resulting from the metal ions binding to the nitrogen lone pairs. The upfield shift of proton h, upon binding of the metal ion, can be explained by a conformational change that moves h out of the deshielding region of the carbonyl oxygens.
 | ||
| Fig. 9
NMR spectra (500 MHz, 297 K) of 4b and its Sr2+ and Ba2+ complexes in CDCl3/MeOD (7:3). | ||
5 Conclusions
In conclusion, we have presented the use of dynamic combinatorial libraries to study hydrazone receptors for metal ions. Small variations in the building block design lead to different molecular recognition properties of the DCLs. Through exploiting a kinetic trap in the system, the receptors could be isolated and characterised. Solution and solid state studies showed that the macrocycles are potent receptors for Sr2+ and Ba2+ while alkali metal ions are not measurably bound. Therefore, hydrazone DCLs provide a useful platform for the discovery of metal ion receptors with unpredictable selectivities. This opens a potential route to new receptors for the extraction of heavy metal ions.Acknowledgements
We thank the Marie-Curie Research Training Network (MRTN-CT-2006-035614, J.M.K. and V.S.), EU (U.L.) and EPSRC (J.K.M.S) for funding, Dr John Davies for solving the X-ray structures, Fabien Cougnon for proof-reading the manuscript and Dr Ana Belenguer for maintaining the HPLC facility.References
- M. Hébrant, Coord. Chem. Rev., 2009, 253, 2186–2192 CrossRef.
- J. R. Bacon and C. M. Davidson, Analyst, 2008, 133, 25–46 RSC.
- G. Dermont, M. Bergeron, G. Mercier and M. Richer-Laflèche, J. Hazard. Mater., 2008, 152, 1–31 CrossRef CAS.
- M. Herrero, J. A. Mendiola, A. Cifuentes and E. Ibánez, J. Chromatogr., A, 2010, 1217, 2495–2511 CrossRef CAS.
- D. Lestan, C. Luo and X. Li, Environ. Pollut., 2008, 153, 3–13 CrossRef CAS.
- J. Peng, Y. Song, P. Yuan, X. Cui and G. Qiu, J. Hazard. Mater., 2009, 161, 633–640 CrossRef CAS.
- B. Singh, J. Hazard. Mater., 2009, 167, 24–37 CrossRef CAS.
- Y. Chen, E. R. M. Mariba, L. V. Dyk and J. H. Potgieter, Int. J. Miner. Process., 2011, 98, 1–7 CrossRef CAS.
- R. S. Forgan, P. A. Wood, J. Campbell, D. K. Henderson, F. E. McAllister, S. Parsons, E. Pidcock, E. M. Swart and P. A. Tasker, Chem. Commun., 2007, 4940–4942 RSC.
- J. M. Klein, V. Saggiomo, L. Reck, M. McPartlin, G. D. Pantos, U. Lüning and J. K. M. Sanders, Chem. Commun., 2011, 47, 3371–3373 RSC.
- S. R. Beeren and J. K. M. Sanders, in Dynamic Combinatorial Chemistry, ed. J. N. H. Reek and S. Otto, Wiley VCH, Weinheim, 2010, ch. 1, pp. 1-22 Search PubMed.
- P. T. Corbett, J. Leclaire, L. Vial, K. R. West, J.-L. Wietor, J. K. M. Sanders and S. Otto, Chem. Rev., 2006, 106, 3652–3711 CrossRef CAS.
- S. Otto, R. L. E. Furlan and J. K. M. Sanders, Science, 2002, 297, 590–593 CrossRef CAS.
- S. Otto and S. Kubik, J. Am. Chem. Soc., 2003, 125, 7804–7805 CrossRef CAS.
- R. T. S. Lam, A. Belenguer, S. L. Roberts, C. Naumann, T. Jarrosson, S. Otto and J. K. M. Sanders, Science, 2005, 308, 667–669 CrossRef CAS.
- P. T. Corbett, J. K. M. Sanders and S. Otto, J. Am. Chem. Soc., 2005, 127, 9390–9392 CrossRef CAS.
- Z. Rodriguez-Docampo, S. I. Pascu, S. Kubik and S. Otto, J. Am. Chem. Soc., 2006, 128, 11206–11210 CrossRef CAS.
- P. T. Corbett, J. K. M. Sanders and S. Otto, Chem.–Eur. J., 2008, 14, 2153–2166 CrossRef CAS.
- R. F. Ludlow and S. Otto, J. Am. Chem. Soc., 2008, 130, 12218–12219 CrossRef CAS.
- R. Perez-Fernandez, M. Pittelkow, A. M. Belenguer, L. A. Lane, C. V. Robinson and J. K. M. Sanders, Chem. Commun., 2009, 3708–3710 RSC.
- V. Saggiomo and U. Lüning, Chem. Commun., 2009, 3711–3713 RSC.
- M. Rauschenberg, S. Bomke, U. Karst and B. J. Ravoo, Angew. Chem., Int. Ed., 2010, 49, 7340–7345 CrossRef CAS.
- S. R. Beeren and J. K. M. Sanders, J. Am. Chem. Soc., 2011, 133, 3804–3807 CrossRef CAS.
- R. L. E. Furlan, Y.-F. Ng, S. Otto and J. K. M. Sanders, J. Am. Chem. Soc., 2001, 123, 8876–8877 CrossRef CAS.
- S. L. Roberts, R. L. E. Furlan, S. Otto and J. K. M. Sanders, Org. Biomol. Chem., 2003, 1, 1625–1633 CAS.
- F. Bulos, S. L. Roberts, R. L. E. Furlan and J. K. M. Sanders, Chem. Commun., 2007, 3092–3093 RSC.
- M.-K. Chung, K. Severin, S. J. Lee, M. L. Waters and M. R. Gagne, Chem. Sci., 2011, 2, 744–747 RSC.
- P. G. Daniele, C. Foti, A. Gianguzza, E. Prenesti and S. Sammartano, Coord. Chem. Rev., 2008, 252, 1093–1107 CrossRef CAS.
- D. D. McRitchie, R. C. Palenik and G. J. Palenik, Inorg. Chim. Acta, 1976, 20, L27–L28 CrossRef CAS.
- C. Lorenzini, C. Pelizzi, G. Pelizzi and G. Predieri, J. Chem. Soc., Dalton Trans., 1983, 721–727 RSC.
- J. Davis and G. J. Palenik, Inorg. Chim. Acta, 1985, 99, L51–L52 CrossRef CAS.
- A. E. Koziol, R. C. Palenik and G. J. Palenik, Inorg. Chim. Acta, 1986, 116, L51–L51 CrossRef CAS.
- A. Bonardi, C. Carini, C. Merlo, C. Pelizzi, G. Pelizzi, P. Tarasconi and F. Vitali, J. Chem. Soc., Dalton Trans., 1990, 2771–2777 RSC.
- M. R. Bermejo, M. Fondo, A. M. Gonzalez-Noya, O. L. Hoyos, A. Sousa, C. A. McAuliffe, W. Hussain, R. Pritchard and V. M. Novotorsev, J. Chem. Soc., Dalton Trans., 1999, 2211–2217 RSC.
- R. Pedrido, M. J. Romero, M. R. Bermejo, A. M. Gonzalez-Noya, M. Maneiro, M. J. Rodriguez and G. Zaragoza, Dalton Trans., 2006, 5304–5314 RSC.
- U. Lüning, R. Baumstark, K. Peters and H. G. von Schnering, Liebigs Ann. Chem., 1990, 129–143 CrossRef.
- M. Hutin, C. J. Cramer, L. Gagliardi, A. R. M. Shahi, G. Bernardinelli, R. Cerny and J. R. Nitschke, J. Am. Chem. Soc., 2007, 129, 8774–8780 CrossRef CAS.
- C. Pelizzi and G. Pelizzi, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1979, 35, 126–128 CrossRef.
- M. R. Bermejo, R. Pedrido, A. M. Gonzalez-Noya, M. J. Romero, M. Vazquez and L. Sorace, New J. Chem., 2003, 27, 1753–1759 RSC.
- U. Lüning, H. Baumgartner and C. Wangnick, Tetrahedron, 1996, 52, 599–604 CrossRef.
- Y. R. Hristova, M. M. J. Smulders, J. K. Clegg, B. Breiner and J. R. Nitschke, Chem. Sci., 2011, 2, 638–641 RSC.
- A. G. Orillo and R. L. E. Furlan, J. Org. Chem., 2010, 75, 211–214 CrossRef.
- P. T. Corbett, S. Otto and J. K. M. Sanders, Chem.–Eur. J., 2004, 10, 3139–3143 CrossRef CAS.
- R. F. Ludlow, J. Liu, H. Li, S. L. Roberts, J. K. M. Sanders and S. Otto, Angew. Chem., Int. Ed., 2007, 46, 5762–5764 CrossRef CAS.
- R. F. Ludlow and S. Otto, Chem. Soc. Rev., 2008, 37, 101–108 RSC.
- J. R. Nitschke, Nature, 2009, 462, 736–738 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic and DCL set up methods, LC-MS, NMR, HRMS and UV-Vis data. CCDC reference numbers 827938 and 827939. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1ob05976a |
| ‡ Present address: University of Groningen, Centre for Systems Chemistry, Stratingh Institute, Nijenborgh 4, 9747 AG Groningen, The Netherlands. |
| This journal is © The Royal Society of Chemistry 2012 |