Towards the systematic exploration of chemical space
Mark
Dow
,
Martin
Fisher
,
Thomas
James
,
Francesco
Marchetti
and
Adam
Nelson
*
Department of Chemistry, University of Leeds, Leeds, UK LS2 9JT
First published on 4th October 2011
Abstract
The discovery of biologically active small molecules is shaped, in large part, by their synthetic (or biosynthetic accessibility). However, chemists' historical exploration of chemical space has been highly uneven and unsystematic. This article describes synthetic strategies that have emerged that may allow chemical space to be explored more systematically. Particular emphasis is placed on approaches that allow the scaffolds of small molecules to be varied combinatorially. In addition, some examples of bioactive small molecules that have been discovered by screening diverse small molecule libraries are highlighted. The authors comment on the likely scope of each of the strategies to deliver skeletally-diverse libraries. In addition, the authors highlight some key challenges for the future: the extension to libraries based on hundreds of distinct scaffolds; and the development of approaches that focus overtly on drug-relevant chemical space.
 Mark Dow | Mark Dow graduated in 2008 from the University of Leicester with a first class MChem degree in Pharmaceutical Chemistry. His PhD, under the supervision of Prof. Adam Nelson and Dr Stuart Warriner, is focussed on the synthesis of a library of skeletally-diverse natural product-like macrocycles. |
 Martin Fisher | Martin Fisher graduated in 2007 from the University of Leeds with a first class MChem degree. Martin began his PhD in 2007 under the supervision of Prof. Adam Nelson and Prof. Colin Fishwick. His project is concerned with the synthesis of structurally diverse antibacterial agents designed using in silico techniques. |
 Thomas James | Thomas James graduated with a first class MChem degree from the University of Leeds in 2009. His studies included an industrial placement with F. Hoffmann-La Roche in Basel, Switzerland. Thomas's PhD project is focussed on the development of methods for the synthesis of diverse, lead-like, heterocycles. |
 Francesco Marchetti | Francesco Marchetti graduated cum laude from the University of Trieste with a research project involving an industrial placement with Eurand. He joined Prof. Roger Griffin's group in 2004 as a PhD student at the Northern Institute for Cancer Research in Newcastle-upon-Tyne. His PhD thesis focussed on the design and synthesis of novel CDK inhibitors as antitumour agents. He is currently a postdoctoral researcher in Prof. Adam Nelson's group working on the synthesis and screening of natural product-like macrocycles. |
 Adam Nelson | Adam Nelson was appointed at the University of Leeds in 1998, and is currently Professor of Chemical Biology and Director of the Astbury Centre for Structural Molecular Biology. Prof. Nelson was awarded the RSC Meldola medal (in 2001), the Pfizer academic award (in 2002), an EPSRC advanced research fellowship (2004–2009), the AstraZeneca award in organic chemistry (in 2005) and the RSC Corday–Morgan medal (in 2008). He has diverse research interests at the interface between chemistry and biology. |
Introduction
Biologically active small molecules continue to make a tremendous contribution to our ability to treat disease,1 and to understand the molecular basis of biological mechanisms.2 The discovery of such molecules is necessarily shaped, in large part, by their synthetic (and biosynthetic) accessibility.3 For example, high-throughput screening requires access to large libraries of small molecules that populate diverse regions of biologically relevant chemical space.4 Even computational approaches,5 for example in which small molecules are docked onto a protein structure,6 require virtual libraries of compounds that can ultimately be either purchased or prepared using established robust synthetic methods. Furthermore, following the initial identification of promising lead molecules (or fragments7), further synthesis is required to drive optimisation.Small molecule scaffolds play a key role in guiding chemists' navigation of biologically-relevant chemical space.8 Vast sources of historic structure–activity relationship data have allowed the chemical space defined by known bioactive ligands to be mapped.9 It has been proposed that biologically active small molecules may be focused in specific sub-fractions of chemical space.10 Indeed, the field of biology-oriented synthesis (BIOS)11 seeks to target such “bioactivity islands” by designing libraries around scaffolds12 that have been biologically validated.
Worryingly, chemists' historical exploration of chemical space using synthesis has been exceptionally uneven and unsystematic. Around half of all known compounds are based on just 0.25% of the known molecular scaffolds (Fig. 1)!13 This uneven exploration is also reflected in small molecule screening collections:14 consequently, the biological properties of small molecules have not been annotated systematically, with huge emphasis on the most synthetically accessible scaffolds. To what extent, then, are our collective views about the biological relevance of chemical space skewed by our uneven exploration by synthesis?
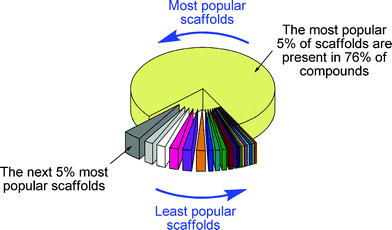 | ||
Fig. 1 Scaffold diversity of the organic chemistry universe. The 24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 282284 cyclic compounds known in the CAS registry in 2008 are grouped from the most popular 5% to the least popular 5% of scaffolds. Around half of known compounds are based on just 0.25% of known molecular scaffolds!13 282284 cyclic compounds known in the CAS registry in 2008 are grouped from the most popular 5% to the least popular 5% of scaffolds. Around half of known compounds are based on just 0.25% of known molecular scaffolds!13 | ||
This article describes synthetic approaches that may allow chemical space to be explored more systematically. A central challenge in this area is to develop synthetic approaches that allow the scaffolds of small molecules to be varied combinatorially. This article will focus on the burgeoning field of diversity-oriented synthesis,15 with specific emphasis on approaches that have emerged to allow control over molecular scaffold.
The “build–couple–pair” approach
An extremely powerful strategy for preparing small molecule libraries with high scaffold diversity involves the exploitation of simple building blocks in combination. The “build–couple–pair” approach requires building blocks to be prepared (“built”) and then connected (“coupled”). Finally, pairs of functional groups are reacted (“paired”) intramolecularly to yield new ring systems in the final scaffolds. The so-called “build–couple–pair” strategy16 has been reviewed.15eThe scope of the “build–couple–pair” strategy is extremely broad, which can be considered to include other diversity-oriented synthetic approaches that have been developed. For example, many ambiphile pairing reactions, folding pathways and branching pathways (see below), as well as all oligomer-based approaches, may be considered to exemplify the “build–couple–pair” strategy.
Ambiphile pairing reactions
A conceptually simple approach to the synthesis of small molecule scaffolds would involve reaction between pairs of bifunctional building blocks;† such reactions have been dubbed “ambiphile pairing reactions” (Fig. 2).17 To be synthetically useful, reactions between the pairs of building blocks need to be selective, and, ideally, can be performed in a single pot. Thus, the initial reaction between the building blocks must be chemoselective (for example, between the pair of functional groups highlighted with circles; Fig. 2); subsequent intramolecular reaction between the remaining functional groups (for example those illustrated with diamonds; Fig. 2) then leads to the formation of a new ring.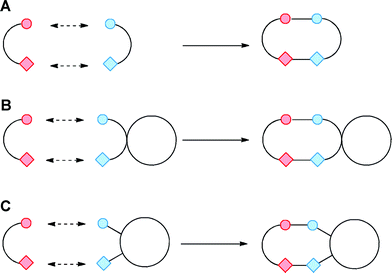 | ||
| Fig. 2 Synthesis of small molecule scaffolds from pairs of bifunctional building blocks using ambiphile pairing reactions. The approach may be used to prepare monocyclic scaffolds (Panel A), or, by using cyclic building blocks, spirocyclic (Panel B) or fused (Panel C) scaffolds. | ||
The overall approach may allow variation of the specific scaffold that is synthesised. First, to vary the functional groups in the new ring, it may be possible to vary the nature of the chemistry used in either the initial condensation or in the cyclisation step. Second, it may be possible to vary the size of the new ring by varying the distance between the reactive functional groups in either building block. Finally, by exploiting cyclic building blocks, polycyclic scaffolds may be prepared (e.g. spirocyclic or fused scaffolds; see Panels B and C, Fig. 2).
Some reactions in which pairs of bifunctional building blocks react selectively to yield new scaffolds are shown in Scheme 1. With Marsden, Nelson has exploited Rh-catalysed Michael additions of o-substituted boronic acid derivatives in the synthesis of a range of heterocycles (Panel A, Scheme 1).18 For example, 2-aminophenyl boronic acids (e.g.1) were reacted with a range of α,β-unsaturated ketones (e.g.2); subsequent cyclisation gave the corresponding cyclic imines, and oxidation yielded the corresponding quinolines (e.g.3).18a It was possible to vary the scaffold prepared by varying the chemistry exploited in the cyclisation step: for example, Rh-catalysed addition of the 2-aminophenyl boronate ester 4 to an α,β-unsaturated ester 5 yielded the corresponding tetrahydroquinolinone 6.18b It is envisaged that the approach may be extended to the synthesis of other heterocyclic scaffolds through further variation of the electrophile and the o-substituted phenylboronic acid derivative used.
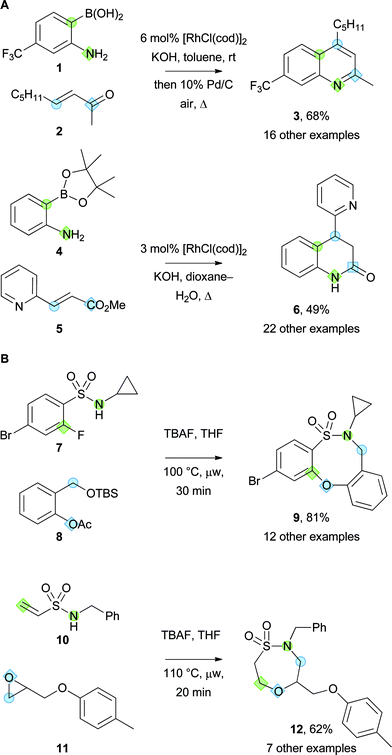 | ||
| Scheme 1 Examples of the synthesis of small molecule scaffolds from bifunctional building blocks using ambiphile pairing reactions. The atoms in different building blocks that ultimately are bonded are highlighted with the same shape. Panel A: Synthesis of heterocyclic scaffolds by reaction between o-substituted boronic acid derivatives and unsaturated carbonyl compounds. Panel B: Syntheses of alternative sultam scaffolds. | ||
A range of sulfonamides with pendant electrophiles has been exploited in the synthesis of sultam scaffolds (Panel B, Scheme 1).17,19 For example, o-fluorobenzenesulfonamides have been reacted with the ortho-quinone methide generated from 8; thus sulfonamides (e.g.7) could be converted into the corresponding eight-membered sultams (e.g.9) with cyclisation by intramolecular SNAr substitution.19 Alternatively, ring-opening of epoxides (e.g.11) with unsaturated sulfonamides (e.g.10) could be followed by oxa-Michael cyclisation to give the corresponding seven-membered sultams (e.g.12).17 It has also been shown that o-fluorobenzenesulfonamides can react selectively with epoxides to give related benzo-fused sultam scaffolds.17 These reactions illustrate a number of tactics to vary the scaffold prepared: the use of cyclic building blocks to give fused scaffolds (e.g.7 to give 9); the variation of the distance between reactive functional groups in building blocks; and the variation of the specific cyclisation chemistry used (compare the syntheses of 9 and 12).
A range of other ambiphile pairing reactions have been developed to allow the synthesis of small molecule scaffolds. For example, organocatalysed reactions between 3-isothiocyanato oxindoles and ketones,20 and between cyclic β-keto amides and unsaturated aldehydes,21 enable the synthesis of spirocyclic scaffolds. In addition, Pd-catalysed approaches have been exploited in 3 + 3 approaches to saturated heterocycles including piperidines.22
Folding and branching pathways
The folding pathway and branching pathway approaches are complementary strategies for the variation of small molecule scaffolds. Folding pathways involve the transformation of alternative substrates into alternative scaffolds under common reaction conditions. In contrast, branching pathways involve the transformation of individual substrates into many different scaffolds under different reaction conditions.Schreiber has developed a folding pathway which exploits the Achmatowicz reaction (Scheme 2).23 The fate of folding pathways depends on strategically placed groups within the substrates (sometimes known as σ-elements).23 The fate of the furans 13 depended on the functionalisation of groups elsewhere within the molecule. Hence, oxidation of the furan 13c, which does not bear any free hydroxy groups, simply generated the enedione 14c. However, with suitably positioned nucleophilic groups in the substrates (as in 13a and 13b), the cis- enedione intermediate could be intercepted to yield either cyclic (as in 14b) or bridged bicyclic (as in 14a) scaffolds. A range of other folding pathways have been developed to yield skeletally diverse small molecules: for example, Rh-catalysed cyclisation–cycloaddition chemistry has been exploited in the synthesis of diverse indole alkaloid-like molecules.24
 | ||
| Scheme 2 A folding pathway based on the Achmatowicz reaction. | ||
The efficiency of folding pathways may be increased if both the construction and the folding of substrates may take place in a single reaction. Within the Nelson group, we have exploited cascade chemistry25—initiated by a three-component reaction—in the synthesis of a range of alkaloid-like scaffolds (Schemes 3).26 Furthermore, the approach could be used in combination with a second three-component reaction to control the substitutional diversity of the final compounds (Scheme 4).
 | ||
| Scheme 3 A cascade reaction involving a folding pathway. After expulsion of molecular nitrogen from 19, 2-aza-dienes 20—the substrates for a folding pathway—are produced. The fate of the reaction of the 2-aza-dienes 20 depends on the substituents R2 and R3 which determine whether 20 is the final product; whether intramolecular Diels–Alder reaction is possible (→ 22 or 23); or whether intramolecular Diels–Alder reaction and subsequent cyclisation takes place (→ 21). | ||
 | ||
| Scheme 4 Selected examples of alkaloid-like compounds that were prepared using two consecutive three-component reactions: a cascade reaction initiated by an inverse-electron demand Diels–Alder reaction; and a Joullié–Ugi reaction. | ||
The reaction between secondary amines 15, carbonyl compounds 16 and triazines 17 yielded a range of skeletally-diverse alkaloid-like compounds. Condensation between the secondary amines 15 and the carbonyl compounds 16 is believed to generate enamines 18 which can undergo an inverse-electron demand Diels–Alder reaction with the triazines 17 (→ 19); expulsion of molecular nitrogen then yields 2-azadienes 20, which are substrates for a subsequent folding pathway. The substituents in 20 determine the molecular scaffold that is ultimately obtained. Thus, in the absence of a tethered dienophile, the 2-aza diene 20 is itself the product of the reaction. However, with an appended dienophile in either the R2 or the R3 substituent, a subsequent intramolecular Diels–Alder reaction can yield either 23 (with R2 = E-octa-3-enyl) or 22 (with R3 = allyl). Finally, with R3 = allyl, and a pendant nucleophile in R2, intramolecular Diels–Alder reaction and subsequent cyclisation is possible (→ 21).
In many cases, the three-component cascade reaction (Scheme 3) could be followed by a second three-component reaction: a Joullié–Ugi reaction. Some selected examples of the alkaloid-like products that were accessible using the approach are shown in Scheme 4. Thus, after the folding pathway, some of the products, such as for example, the 2-aza diene 20a, the cyclic imines 22a, 22b and 23a, were reacted with an isocyanide and a carboxylic acid to give final products (such as 24–27). In contrast, with an appropriate pendant nucleophile in place (such as the NHCbz group in 16e), a cyclisation terminated the three-component cascade reaction (→21a). In total, 43 final products were prepared which were, in general derived from five separate components. The products were based on 28 distinct graph-node-level frameworks:‡ the high scaffold diversity of the products stemmed both from rings found in the individual components and from the power of the folding pathway.
Oguri and colleagues demonstrated a powerful strategy utilising metathesis to ‘fold’ alternative substrates into polycyclic ring systems (Scheme 5).27 The folding substrates 29a–f were prepared in 3–4 steps from the α,β-unsaturated nitrile 28. Subsequent folding, using ring-closing ene–yne–ene metathesis cascade chemistry, generated the tricyclics 30a–f. Further synthetic transformations yielded analogues of the sesquiterpene artemisinin (34), some of which exhibited anti-trypanosomal activity.
 | ||
| Scheme 5 A folding pathway leading to analogues of the sesquiterpene artemisinin. | ||
In contrast, branching pathways involve the conversion of common precursors into a range of distinct molecular scaffolds through careful choice of the reaction conditions. An ingenious branching pathway, which exploits complementary cyclisation reactions, is illustrated in Scheme 6.28 A four-component Petasis reaction was used to assemble flexible cyclisation precursors (e.g.35). Alternative cyclisation reactions were then used to yield products with distinct molecular scaffolds: Pd-catalysed cyclisation (→ 36a); enyne metathesis (→ 36b); Ru-catalysed cycloheptatriene formation (→ 36c); Au-catalysed cyclisation of an alcohol onto an alkyne (→ 36d); base-induced cyclisation (→ 36e); Pauson–Khand reaction (→ 36f); and [2,3]-sigmatropic rearrangement (not shown). Four of these cyclisation reactions could be used again to convert the enyne 36e into molecules with four further scaffolds (37b–e). In addition dienes (such as 36b) were substrates for Diels–Alder reactions (to yield adducts such as 37a). The key to the approach lay in design of precursors (e.g.35) which were effective substrates for a range of efficient and diastereoselective cyclisation reactions. Other successful branching pathways have also exploited complementary transition metal-catalysed cyclisation reactions.29
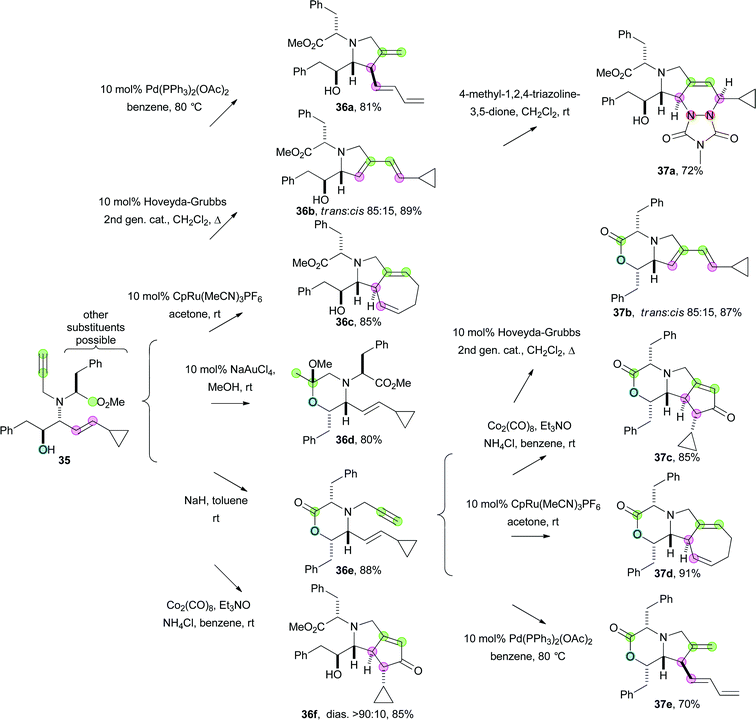 | ||
| Scheme 6 A branching pathway exploiting complementary cyclisation reactions. | ||
Spring has developed a branching pathway which exploits numerous, asymmetric, reactions (Scheme 7).30 This approach utilises the diverse reactivity of α,β-unsaturated acyl imidazolidinone 39. The unsaturated imidazolidinone 39 underwent 1,3-dipolar cycloaddition (→ 40), asymmetric dihydroxylation (→ 41) and catalytic, asymmetric Diels–Alder (→ 42) reaction. From this initial branching point, the diverse functional groups embedded within 40, 41 and 42 were exploited in further branching pathways, for example, metathesis cascades (→ 46c) and oxidative cleavage followed by tandem reductive amination (→ 47a). This synthetic approach enabled the discovery of gemmacin (based on the framework 47a) which exhibits antibacterial activity towards methicillin-resistant Staphylococcus aureus (MRSA).
 | ||
Scheme 7 Branching pathway based on complementary catalytic asymmetric reactions. Reagents and conditions: (a) LiBr, DBU, R1CHO, MeCN; (b) (R)-QUNIAP, AgOAc, iPr2NEt, THF, −78 → 25 °C; (c) AD-mix, DHQD2PHAL, 1:1 THF–H2O; (d) chiral bisoxazoline ligand, Cu(OTf)2, 3 Å sieves, CH2Cl2; (e) R2COCl, DMAP, pyridine, CH2Cl2; (f) R3CHO, BH3·pyridine, MeOH; (g) SOCl2, pyridine, CH2Cl2, 40 °C; (h) R4Br, Ag2O, CH2Cl2, 40 °C; (i) R5C(O)R5, TsOH, DMF, 65 °C; (j) R6CHO, TsOH, DMF, 65 °C; (k) NaN3, DMF, 100 °C then dimethyl acetylenedicarboxylate, PhMe, 65 °C; (l) mCPBA, CH2Cl2 then MeOH, 65 °C; (m) CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCO2Bn, ethylene, cat. Grubbs II catalyst, PhMe, 120 °C; (n) OsO4, NMO, 10:1 acetone–H2O; (o) RNH2, Me2AlCl, PhMe, 120 °C; then NaH, R11X, DMF, THF; then PhMe, cat. Grubbs II catalyst, ethylene, 120 °C; (p) NaIO4, 1:1 THF–H2O; then R7NH2, NaB(OAc)3H, CH2Cl2; (q) NaIO4, 1:1 THF–H2O then R8NHR8, NaB(OAc)3H, CH2Cl2; (r) R9CHO, DMF, TsOH, 60 °C; (s) R10C(O)R10, DMF, TsOH, 60 °C. CHCO2Bn, ethylene, cat. Grubbs II catalyst, PhMe, 120 °C; (n) OsO4, NMO, 10:1 acetone–H2O; (o) RNH2, Me2AlCl, PhMe, 120 °C; then NaH, R11X, DMF, THF; then PhMe, cat. Grubbs II catalyst, ethylene, 120 °C; (p) NaIO4, 1:1 THF–H2O; then R7NH2, NaB(OAc)3H, CH2Cl2; (q) NaIO4, 1:1 THF–H2O then R8NHR8, NaB(OAc)3H, CH2Cl2; (r) R9CHO, DMF, TsOH, 60 °C; (s) R10C(O)R10, DMF, TsOH, 60 °C. | ||
Kozmin reported a strategy which produced a library of nearly 200 members with a broad distribution of molecular shapes and physicochemical properties (Scheme 8).31 The approach was based upon an initial branching pathway from the enyne 48, utilising a variety of transition metal mediated cycloisomerisations, leading to 1,3-dienes (49a → 49d). 1,3-Dienes were reacted combinatorially with five dienophiles, generating a diverse array of mono- and polycyclic compounds (e.g.50d and 50c). The Diels–Alder products were dihydroxylated to yield the corresponding diols with good to excellent diastereoselectivity. Finally, tin-mediated mono- or bis-carbamylation of diols, completed the synthesis of the library. Screening of the library led to the identification of a compound, 52, which suppressed glycolytic production of ATP and lactate in CHO-K1 cell lines.
 | ||
| Scheme 8 A branching pathway emanating from a common enyne starting material. | ||
Oligomer-based approaches to scaffold diversity
A powerful version of the “build–couple–pair” strategy involves the iterative assembly, and subsequent cyclisation, of oligomeric substrates.33 An oligomer-based approach has been used to harness the rich cyclisation chemistry of N-acyl iminium ions (Scheme 9).32 Initially, peptide synthesis was used to prepare peptides 55 which contained a masked aldehyde, a suitably positioned secondary amide, and a pendant nucleophile. Treatment of the peptides with acid triggered the release of an aldehyde, the formation of an N-acyl iminium ion, and the cyclisation to yield final scaffolds (e.g.56–61).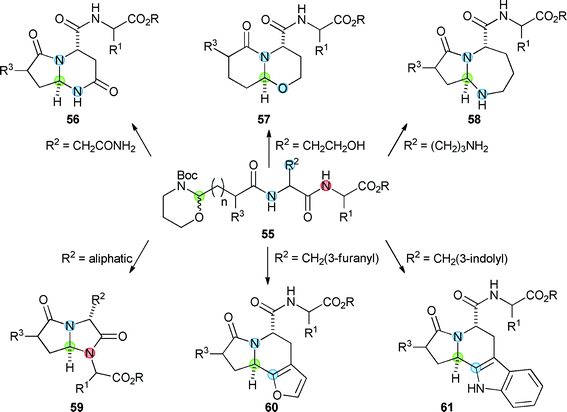 | ||
| Scheme 9 Acid-catalysed cyclisation of N-acyl iminium ions to yield a range of small molecule scaffolds. | ||
Metathesis cascades involving oligomeric substrates have underpinned the synthesis of skeletally diverse small molecule libraries.32,34 Using this approach, we have, within the Nelson group, prepared natural product-like molecules with unprecedented scaffold diversity (Scheme 10).34 A wide range of oligomeric metathesis substrates were prepared by the iterative attachment of unsaturated building blocks to a fluorous-tagged linker. Crucially, alternative attachment reactions were used such that the building blocks were connected through bonds that either did, or did not, remain as a vestige in the final products. Finally, metathesis cascades were used to “reprogramme” the scaffolds, and to release the products of successful cascade processes from the fluorous tag. The resulting library had unprecedented scaffold diversity (over eighty distinct molecular scaffolds).
 | ||
| Scheme 10 Synthesis of natural product-like molecules with unprecedented scaffold diversity. Oligomeric metathesis substrates were assembled by iterative attachment of “propagating” and “terminating” building blocks to a fluorous-tagged linker via either temporary or permanent linkages; the functional groups used to oligomerise the building blocks are highlighted with diamonds. The molecular scaffolds were reprogrammed through metathesis cascade reactions (between unsaturated functional groups highlighted with circles), with concomitant release from the fluorous tag. After deprotection, small molecules with over eighty distinct molecular scaffolds were obtained. X = OH or NHNs. | ||
We extended the approach by exploiting metathesis cascade chemistry in combination with either inter- or intramolecular Diels–Alder reactions (Scheme 11).35 The metathesis substrates (e.g.71–73) were assembled iteratively from the corresponding building blocks (in the case of 71 and 72, using a fluorous tagged “safety catch” linker that we had previously developed36). In the illustrated cases, the metathesis cascade resulted in the formation of a 1,3-diene whose reactivity was exploited in a Diels–Alder reaction. The fate of the 1,3-dienes 74–76 illustrates three distinct tactics that we employed: attachment of a dienophile and intramolecular Diels–Alder reaction (→ 77); intermolecular Diels–Alder reaction (→ 78); and intramolecular Diels–Alder reaction involving a tethered dienophile also formed in the cascade (→ 79).
 | ||
| Scheme 11 Exploitation of metathesis cascades in combination with inter- and intramolecular Diels–Alder reactions. Metathesis substrates were assembled iteratively from building blocks, and subjected to metathesis cascade reactions. In the illustrated cases, the cascade resulted in the formation of a 1,3-diene whose reactivity was exploited in a Diels–Alder reaction. | ||
Summary and outlook
The historically uneven exploration of chemical space using synthesis presents a huge challenge to synthetic chemists: to develop synthetic approaches that allow chemical space to be probed more systematically. This challenge has required the development of synthetic approaches that allow molecule scaffolds to be varied combinatorially. The development of the “branching” and “folding” pathway strategies has enabled the synthesis of libraries based on up to ∼30 scaffolds. These strategies are extremely ingenious; however, extension to the libraries based on many scores of alternative scaffolds is likely to be difficult (and unlikely to be general).How feasible will it be, then, to devise reliable syntheses of molecules based on hundreds or even thousands of distinct scaffolds? Oligomer-based approaches provide the first glimpse into how this challenge might be met. The assembly of oligomeric substrates iteratively, using reliable reactions, may be combined with cyclisation reactions of broad scope. A significant challenge will be to identify reactions other than metathesis that have the broad scope and chemoselectivity needed to yield molecular scaffolds combinatorially. But it is at least possible that oligomer-based approaches may, in the future, be harnessed to yield libraries that target huge swathes of chemical space.
Another major challenge is to develop diversity-oriented synthetic methods that map onto the requirements of drug discovery programmes.37 Established diversity-oriented approaches have tended to focus on small molecules that lie well outside drug-like space. How, then, might these approaches be adapted to be more relevant in drug discovery? It is now generally accepted that attrition rates are strongly linked to molecular properties38 such as molecular weight, lipophilicity, the number of aromatic rings,38a and the fraction of sp3-hybridised38b carbon atoms. Optimisation almost always leads to increases in both molecular weight and lipophilicity. So it is essential to have good starting points for optimisation, and, therefore, to control the properties of initial leads. The development of general strategies that are able to deliver skeletally diverse compounds—but within the boundaries of lead-like space—is likely to be extremely demanding. But this goal is, nonetheless, an important challenge for synthetic chemists in the twentyfirst century.
Acknowledgements
We thank EPSRC, AstraZeneca and GSK for supporting our current research in this area, and Dr Stuart Warriner for helpful discussions.References
- (a) Drug Discovery and Development. Volume 1: Drug Discovery, ed. M. S. Chorghade, Wiley, Hoboken, 2006 Search PubMed; (b) Accounts in Drug Discovery. Case Studies in Medicinal Chemistry, ed. J. C. Barrish, P. H. Carter, P. T. W. Cheng and R. Zahler, Royal Society of Chemistry, Cambridge, 2011 Search PubMed.
- (a) M. Fisher and A. Nelson, in New Frontiers in Chemical Biology, ed. M. E. Bunnage, Royal Society of Chemistry, Cambridge, 2011, ch. 1 Search PubMed; (b) D. P. Walsh and Y. T. Chang, Chem. Rev., 2006, 106, 2476–2530 CrossRef CAS.
- (a) M. A. Koch, A. Schuffenhauer, M. Scheck, S. Wetzel, M. Casaulta, A. Odermatt, P. Ertl and H. Waldmann, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 17272–17277 CrossRef CAS; (b) M. A. Koch, L. O. Wittenberg, S. Basu, D. A. Jeyaraj, E. Gourzoulidou, K. Reinecke, A. Odermatt and H. Waldmann, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 16721–16726 CrossRef CAS; (c) C. Lipinski and A. Hopkins, Nature, 2004, 432, 855–861 CrossRef CAS.
- (a) High-Throughput Screening in Drug Discovery. Methods and Principles in Medicinal Chemistry, ed. J. Hüser, Wiley-VCH, Weinheim, 2007, vol. 35 Search PubMed; (b) K. H. Bleicher, H. J. Böhm, K. Müller and A. I. Alanine, Nat. Rev. Drug Discovery, 2003, 2, 369–378 CrossRef CAS.
- For examples in the discovery of antibacterials, see: K. J. Simmons, I. Chopra and C. W. G. Fishwick, Nat. Rev. Microbiol., 2010, 8, 501–510 CrossRef CAS.
- D. B. Kitchen, H. Decornez, J. R. Furr and J. Bajorath, Nat. Rev. Drug Discovery, 2004, 3, 935–949 CrossRef CAS.
- (a) D. C. Rees, M. Congreve, C. W. Murray and R. Carr, Nat. Rev. Drug Discovery, 2004, 3, 660–672 CrossRef CAS; (b) P. J. Hajduk and J. Greer, Nat. Rev. Drug Discovery, 2007, 6, 211–219 CrossRef CAS.
- (a) S. Wetzel, K. Klein, S. Renner, D. Rauh, T. I. Oprea, P. Mutzel and H. Waldmann, Nat. Chem. Biol., 2009, 5, 581–583 CrossRef CAS; (b) S. Renner, W. A. L. van Otterlo, M. D. Seoane, S. Möcklinghoff, B. Hofmann, S. Wetzel, A. Schuffenhauer, P. Ertl, T. I Oprea, D. Steinhilber, L. Brunsveld, D. Rauh and H. Waldmann, Nat. Chem. Biol., 2009, 5, 585–592 CrossRef CAS.
- (a) A. A. Shelat and R. K. Guy, Nat. Chem. Biol., 2007, 3, 442–446 CrossRef CAS; (b) Y. Hu, A. Mai Wassermann, E. Lounkine and J. Bajorath, J. Med. Chem., 2010, 53, 752–758 CrossRef CAS; (c) J J. Hert, J. J. Irwin, C. Laggner, M. J. Keiser and B. K. Shoichet, Nat. Chem. Biol., 2009, 5, 479–483 CrossRef CAS; (d) G. V. Paolini, R. H. B. Shapland, W. P. van Hoorn, J. S. Mason and A. L. Hopkins, Nat. Biotechnol., 2006, 24, 805–815 CrossRef CAS.
- H. Waldmann, Nat. Chem. Biol., 2009, 5, 76–77 CrossRef CAS.
- A. Nören-Müller, I. Reis-Correa, H. Prinz, C. Rosenbaum, K. Saxena, H. J. Schwalbe, D. Vestweber, G. Cagna, S. Schunk, O. Schwarz, H. Schiewe and H. Waldmann, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10606–10611 CrossRef.
- For the concept of a privileged scaffold, see: B. E. Evans, K. E. Rittle, M. G. Bock, R. M. DiPardo, R. M. Freidinger, W. L. Whitter, G. F. Lundell, D. F. Veber and P. S. Anderson, J. Med. Chem., 1988, 31, 2235–2246 CrossRef CAS.
- A. H. Lipkus, Q. Yuan, K. A. Lucas, S. A. Funk, W. F. Bartelt III, R. J. Schenck and A. J. Trippe, J. Org. Chem., 2008, 73, 4443–4451 CrossRef CAS.
- For the assessment of scaffold diversity in compound libraries, see: M. Krier, G. Bret and D. Rognan, J. Chem. Inf. Model., 2006, 46, 512–524 CrossRef CAS.
- (a) M. D. Burke and S. L. Schreiber, Angew. Chem., Int. Ed., 2004, 43, 46–58 CrossRef; (b) W. R. J. D. Galloway, A. Isidro-Llobet and D. R. Spring, Nat. Commun., 2010, 1, 1–13 CrossRef; (c) S. Dandapani and L. A. Macaurelle, Curr. Opin. Chem. Biol., 2010, 14, 362–370 CrossRef CAS; (d) C. Cordier, D. Morton, S. Murrison, A. Nelson and C. O'Leary-Steele, Nat. Prod. Rep., 2008, 25, 719–737 RSC; (e) T. E. Nielsen and S. L. Schreiber, Angew. Chem., Int. Ed., 2008, 47, 48–56 CrossRef CAS.
- T. Uchida, M. Rodriquez and S. L. Schreiber, Org. Lett., 2009, 11, 1559–1562 CrossRef CAS.
- A. Rolfe, T. B. Samarakoon and P. R. Hanson, Org. Lett., 2010, 12, 1216–1219 CrossRef CAS.
- (a) J. Horn, S. P. Marsden, A. Nelson, D. House and G. G. Weingarten, Org. Lett., 2008, 10, 4117–4120 CrossRef CAS; (b) J. Horn, H. Y. Li, S. P. Marsden, A. Nelson, R. J. Shearer, A. J. Campbell, D. House and G. G. Weingarten, Tetrahedron, 2009, 65, 9002–9007 CrossRef CAS.
- T. B. Samarakoon, M. Y. Hur, R. D. Kurtz and P. R. Hanson, Org. Lett., 2010, 12, 2182–2185 CrossRef CAS.
- W. Chen, Z. Wu, J. Hu, L. F. Cun, X. M. Zhang and W. C. Yuan, Org. Lett., 2011, 13, 2472–2475 CrossRef CAS.
- M. del Mar Sanchez Duque, O. Basl, N. Isambert, A. Gaudel-Siri, Y. Gnisson, J. C. Plaquevent, J. Rodriguez and T. Constantieux, Org. Lett., 2011, 13, 3296–3299 CrossRef.
- (a) K. M. Goodenough, P. Raubo and J. P. A. Harrity, Org. Lett., 2005, 7, 2993–2996 CrossRef CAS; (b) S. J. Hedley, W. J. Moran, D. A. Price and J. P. A. Harrity, J. Org. Chem., 2003, 68, 4286–4292 CrossRef CAS.
- (a) M. D. Burke, E. M. Berger and S. L. Schreiber, J. Am. Chem. Soc., 2004, 126, 14095–14104 CrossRef CAS; (b) M. D. Burke, E. M. Berger and S. L. Schreiber, Science, 2003, 302, 613–618 CrossRef CAS.
- H. Oguri and S. L. Schreiber, Org. Lett., 2005, 7, 47–50 CrossRef CAS.
- (a) S. Murrison, D. Glowacki, C. Einzinger, J. Titchmarsh, S. Bartlett, B. McKeever-Abbas, S. Warriner and A. Nelson, Chem.–Eur. J., 2009, 15, 2185–2189 CrossRef CAS; (b) W. J. Bromley, M. Gibson, S. Lang, S. A. Raw, A. C. Whitwood and R. J. K. Taylor, Tetrahedron, 2007, 63, 6004–6014 CrossRef CAS; (c) S. A. Raw and R. J. K. Taylor, J. Am. Chem. Soc., 2004, 126, 12260–12261 CrossRef CAS.
- S. Murrison, S. K. Maurya, C. Einzinger, B. McKeever-Abbas, S. Warriner and A. Nelson, Eur. J. Org. Chem., 2011, 2354–2359 CrossRef CAS.
- H. Oguri, T. Hiruma, Y. Yamagishi, H. Oikawa, A. Ishiyama, K. Otoguro, H. Yamada and S. Omura, J. Am. Chem. Soc., 2011, 133, 7096–7105 CrossRef CAS.
- N. Kumagai, G. Muncipinto and S. L. Schreiber, Angew. Chem., Int. Ed., 2006, 45, 3635–3638 CrossRef CAS.
- G. Moura-Letts, C. M. DiBlasi, R. A. Bauer and D. S. Tan, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6745–6750 CrossRef CAS.
- G. L. Thomas, R. J. Spandl, F. G. Glansdorp, M. Welch, A. Bender, J. Cockfield, J. A. Lindsay, C. Bryant, D. F. J. Brown, O. Loiseleur, H. Rudyk, M. Ladlow and D. R. Spring, Angew. Chem., Int. Ed., 2008, 47, 2808–2812 CrossRef CAS.
- J. Cui, J. Hao, O. A. Ulanovskaya, J. Dundas, J. Liang and S. A. Kozmin, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6763–6768 CrossRef CAS.
- D. A. Spiegel, F. C. Schroeder, J. R. Duvall and S. L. Schreiber, J. Am. Chem. Soc., 2006, 128, 14766–14767 CrossRef CAS.
- (a) T. E. Nielsen and M. Meldal, J. Org. Chem., 2004, 69, 3765 CrossRef CAS; (b) T. E. Nielsen and M. Meldal, J. Comb. Chem., 2005, 7, 599 CrossRef CAS; (c) T. E. Nielsen, S. L. Quement and M. Meldal, Org. Lett., 2005, 7, 3601 CrossRef CAS.
- D. Morton, S. Leach, C. Cordier, S. Warriner and A. Nelson, Angew. Chem., Int. Ed., 2009, 48, 104–109 CrossRef CAS.
- C. O'Leary-Steele, P. J. Pedersen, T. James, T. Lanyon-Hogg, S. Leach, J. Hayes and A. Nelson, Chem.–Eur. J., 2010, 16, 9563–9571 CrossRef CAS.
- C. O'Leary-Steele, C. Cordier, J. Hayes, S. Warriner and A. Nelson, Org. Lett., 2009, 11, 915–918 CrossRef CAS.
- The range of reactions widely used in array synthesis is rather limited: (a) T. W. J. Cooper, I. B. Campbell and S. J. F. Macdonald, Angew. Chem., Int. Ed., 2010, 49, 8082–8091 CrossRef CAS; (b) S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451–3479 CrossRef CAS.
- (a) T. J. Ritchie and S. J. F. Macdonald, Drug Discovery Today, 2009, 14, 1011–1020 CrossRef CAS; (b) F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS; (c) G. Keserü and G. M. Makara, Nat. Rev. Drug Discovery, 2009, 8, 203–212 CrossRef; (d) P. D. Leeson and B. Springthorpe, Nat. Rev. Drug Discovery, 2007, 6, 881–890 CrossRef CAS; (e) M. C. Wenlock, R. P. Austin, P. Barton, A. M. Davis and P. D. Leeson, J. Med. Chem., 2003, 46, 1250–1256 CrossRef CAS.
Footnotes |
| † Throughout this article, the reactive atoms in each building block are highlighted in the same colour. In some schemes, the atoms in different building blocks that ultimately become bonded are highlighted by the same shape. |
| ‡ Frameworks defined at the graph-node level describe the connectivity and type of the atoms (but not the bond types between atoms). |
| This journal is © The Royal Society of Chemistry 2012 |