A concise asymmetric synthesis of (−)-rasfonin†
Yange
Huang
,
Adriaan J.
Minnaard
* and
Ben L.
Feringa
*
Stratingh Institute for Chemistry, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands. E-mail: a.j.minnaard@rug.nl; b.l.feringa@rug.nl; Fax: +31 50 363 4278; Tel: +31 50 363 4296
First published on 24th November 2011
Abstract
A very efficient total synthesis of the apoptosis inducer (−)-rasfonin has been developed using CuBr/JosiPhos catalyzed iterative asymmetric conjugate addition of MeMgBr and Feringa's butenolide.
Natural products containing α-pyranones (δ-lactones) show a variety of interesting biological properties.1 As a prime example, rasfonin 1, isolated from the fungus Trichurus terrophilus2 and the fermented mycelium of Taleromyces species 3565-A1,3 has been reported as an active apoptosis inducer in ras-dependent cells.4 In connection with this finding, significant proliferation suppression of mouse splenic lymphocytes stimulated with mitogens, concanavalin A and lipopolysaccharide, was reported.4
Due to the potential use of (−)-1 in the development of cancer chemotherapeutics, a versatile synthetic route to rasfonin is required to establish which parts of the molecule are important for activity, and to pin down its target protein(s). The first total synthesis of (−)-1, reported by Ishibashi and co-workers in 2003, aimed at structure elucidation and absolute configuration determination.2,5 A second synthesis, reported by the group of Boeckman in 2006,6 was based on the use of camphor lactam chiral auxiliaries in order to allow the synthesis of different stereoisomers. As no follow-up appeared in chemical biology, we felt that a concise synthesis of 1, taking advantage of highly efficient and selective catalytic methods and making this compound and analogs more readily available, could greatly stimulate biological studies. Herein, we report the asymmetric synthesis of (−)-1 in a highly efficient and selective manner.
In our retrosynthetic analysis (Scheme 1), (−)-rasfonin 1 was disconnected into upper half 2 and lower half 3. The former was planned to be obtained from 4, in turn prepared via our iterative catalytic asymmetric conjugate addition protocol to deoxypropionates,7 in combination with a stereospecific Achmatowicz rearrangement. The lower part, 3, should in principle be accessible starting from readily available enantiopure Feringa's menthyl butenolide 5.8
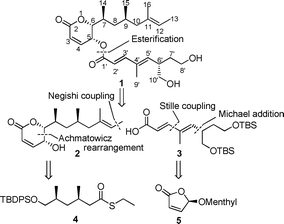 | ||
| Scheme 1 Retrosynthesis of (−)-rasfonin 1. | ||
Syn-1,3-dimethyl thioester
4 was synthesized in an excellent 57% overall yield starting from ethylene glycol 6 by CuBr/JosiPhos catalyzed iterative asymmetric conjugate addition of MeMgBr (Scheme 2).9 Mono-protection of ethylene glycol 6 gave alcohol 7. IBX oxidation of alcohol 7 followed by Wittig reaction using Wittig reagent 18 afforded substrate 9 in 86% yield. Substrate 9 gave excellent yield and enantioselectivity (95%, 95% ee) of compound 10 using CuBr/JosiPhos catalyzed conjugate addition of MeMgBr. DIBAL reduction of compound 10 followed by HWE olefination using reagent 19 gave thioester 11 in 89% yield. The excellent syn selectivity (96![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4) of the second asymmetric conjugate addition of MeMgBr catalyzed by CuBr/JosiPhos gave dimethyl thioester 4 in 96% yield. Reduction of 4 with DIBAL afforded the corresponding aldehyde which was used immediately to form the terminal alkyne 12 by addition of lithiated trimethylsilyldiazomethane involving a Colvin rearrangement.10Vinyl iodide 13 was subsequently prepared by Zr-catalyzed methylalumination,11 followed by Negishi cross coupling12 with ZnMe2 to afford alkene 14, which after deprotection with TBAF provided alcohol 15 in 87% yield. Ley oxidation13 of 15 afforded the corresponding aldehyde which was treated with 2-furyl lithium to give the corresponding furyl alcohol in 88% yield (syn:anti = 2:1). Subsequent Ley oxidation of this mixture gave ketone 16 in 98% yield which was in turn treated with (S)-CBS reagent and borane14 to afford furyl alcohol 17 in an excellent 94% yield and a syn:anti ratio >98:2. Stereospecific Achmatowicz rearrangement15 of 17 with vanadyl acetylacetonate and tert-butylhydroperoxide afforded the hemi-acetal in 69% yield which was subsequently oxidized by Jones' reagent16 to the corresponding ketolactone. Finally, Luche reduction17 gave the upper half 2 as the only diastereomer.
:4) of the second asymmetric conjugate addition of MeMgBr catalyzed by CuBr/JosiPhos gave dimethyl thioester 4 in 96% yield. Reduction of 4 with DIBAL afforded the corresponding aldehyde which was used immediately to form the terminal alkyne 12 by addition of lithiated trimethylsilyldiazomethane involving a Colvin rearrangement.10Vinyl iodide 13 was subsequently prepared by Zr-catalyzed methylalumination,11 followed by Negishi cross coupling12 with ZnMe2 to afford alkene 14, which after deprotection with TBAF provided alcohol 15 in 87% yield. Ley oxidation13 of 15 afforded the corresponding aldehyde which was treated with 2-furyl lithium to give the corresponding furyl alcohol in 88% yield (syn:anti = 2:1). Subsequent Ley oxidation of this mixture gave ketone 16 in 98% yield which was in turn treated with (S)-CBS reagent and borane14 to afford furyl alcohol 17 in an excellent 94% yield and a syn:anti ratio >98:2. Stereospecific Achmatowicz rearrangement15 of 17 with vanadyl acetylacetonate and tert-butylhydroperoxide afforded the hemi-acetal in 69% yield which was subsequently oxidized by Jones' reagent16 to the corresponding ketolactone. Finally, Luche reduction17 gave the upper half 2 as the only diastereomer.
 | ||
| Scheme 2 Synthesis of upper half of (−)-rasfonin 1. | ||
The stereogenic center in the lower half of (−)-rasfonin was introduced by Michael addition of lithium bis(phenylthio)methane8 to butenolide 5 to provide trans-20 as the single diastereomer in 86% yield (Scheme 3). Full reduction of 20 by LiAlH4 afforded diol 21 in 90% yield which after protection provided 22 in 96% yield. Unmasking of dithiane 22 by HgCl2 and HgO in a mixture of acetonitrile and water18 went smoothly and afforded the free aldehyde in 85% yield which upon treatment with lithiated trimethylsilyldiazomethane provided the terminal alkyne 23 in 67% yield. Terminal alkylation of the alkyne moiety to give 24 was performed by lithiation followed by quenching with methyl iodide. The subsequent Pd-catalyzed hydrostannation of alkyne 24 with catalytic Pd(PPh3)Cl2 initially gave low yield and incomplete conversion. We encountered this problem before,19 and again the method proposed by Semmelhack and Hooley20 strongly improved the outcome. Switching to catalytic Pd(OAc)2 and tricyclohexyl phosphine, with hexane as the solvent, led to complete conversion and 80% yield in 20 min! Vinyl iodide 25 was subsequently obtained in 75% yield by treating this stannylated compound with iodine. Stille coupling of acid 2621 and 25 provided lower half 3 in 87% yield.
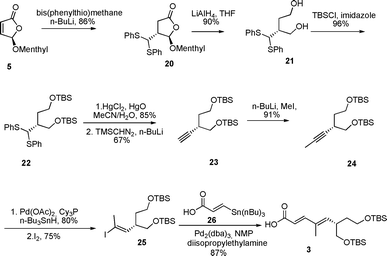 | ||
| Scheme 3 Synthesis of the lower half of (−)-rasfonin 1. | ||
The coupling of the upper half 2 and lower half 3 of (−)-rasfonin was achieved by Yamaguchi esterification22 in 80% yield (Scheme 4). Desilylation initially was not satisfactory as treatment with camphorsulfonic acid gave only 40% yield.6 Fortunately, switching to aq. HF in acetonitrile23 gave in a close to quantitative yield (−)-rasfonin whose optical rotation and spectroscopic data agreed with the reported values (except for the presence of approx. 5% of a diastereomer that could not be separated).
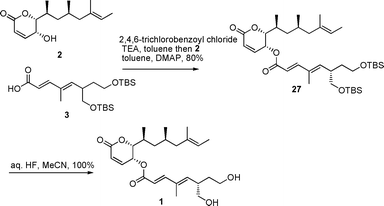 | ||
| Scheme 4 Coupling of the upper half and lower half to give (−)-rasfonin 1. | ||
In conclusion, a very efficient total synthesis of the apoptosis inducer (−)-rasfonin has been developed. CuBr/JosiPhos catalyzed iterative asymmetric conjugate addition of MeMgBr has been employed to install the stereogenic centers in the upper half side chain with excellent yield and stereoselectivity. The hydroxy-lactone core could be prepared by a subsequent stereospecific hydroxy-directed Achmatowicz rearrangement followed by an oxidation–reduction sequence. The synthesis of the lower half 3 makes use of the perfect transfer of chirality in the conjugate addition to butenolide 5 followed by selective construction of the E,E-diene-ester part. The availability of an effective route to rasfonin now allows us to study its role in inhibiting the Ras signalling pathway, provides access to functional analogs and might lead to the identification of its target protein.
Acknowledgements
T. D. Tiemersma-Wegman (Stratingh Institute for Chemistry) is acknowledged for chromatography support. Financial support from NRSC catalysis grant 200910018B is gratefully acknowledged.Notes and references
- (a) V. Shekhar, D. K. Reddy, V. Suresh, D. C. Babu and Y. Venkateswarlu, Tetrahedron Lett., 2010, 51, 946 CrossRef CAS; (b) A. Favre, F. Carreaux, M. Deligny and B. Carboni, Eur. J. Org. Chem., 2008, 4900 Search PubMed; (c) L. Dong, V. A. Gordon, R. L. Grange, J. Johns, P. G. Parsons, A. Porzelle, P. Reddell, H. Schill and C. M. Williams, J. Am. Chem. Soc., 2008, 130, 15262 Search PubMed.
- K. Akiyama, S. Kawamoto, H. Fujimoto and M. Ishibashi, Tetrahedron Lett., 2003, 44, 8427 Search PubMed.
- T. Tomikawa, K. Shin-Ya, K. Furihato, T. Kinoshita, A. Miyajima, H. Seto and Y. Hayakawa, J. Antibiot., 2000, 53, 848 Search PubMed.
- (a) H. Fujimoto, E. Sone, E. Okuyama, M. Ishibashi, 120th Annual Meeting of the Pharmaceutical Society of Japan, Abstracts of Papers 2, 2000; p 68; (b) O. Rocks, A. Peyker, M. Kahms, P. J. Verveer, C. Koerner, M. Lumbierres, J. Kuhlmann, H. Waldmann, A. Wittinghofer and P. I. H. Bastiaens, Science, 2005, 307, 1746 CrossRef CAS; (c) L. Brunsveld, J. Kuhlmann, K. Alexandrov, A. Wittinghofer, R. S. Goody and H. Waldmann, Angew. Chem., Int. Ed., 2006, 45, 6622 CrossRef CAS; (d) F. J. Dekker, O. Rocks, N. Vartak, S. Menninger, C. Hedberg, R. Balamurugan, S. Wetzel, S. Renner, M. Gerauer, B. Scholermann, M. Rusch, J. W. Kramer, D. Rauh, G. W. Coates, L. Brunsveld, P. I. H. Bastiaens and H. Waldmann, Nat. Chem. Biol., 2010, 6, 449 Search PubMed.
- K. Akiyama, S. Yamamoto, H. Fujimoto and M. Ishibashi, Tetrahedron, 2005, 61, 1827 CrossRef CAS.
- R. K. Boeckman Jr., J. E. Pero and D. J. Boehmler, J. Am. Chem. Soc., 2006, 128, 11032 Search PubMed.
- (a) R. D. Mazery, M. Pullez, F. López, S. R. Harutyunyan, A. J. Minnaard and B. L. Feringa, J. Am. Chem. Soc., 2005, 127, 9966 CrossRef CAS; (b) B. ter Horst, B. L. Feringa and A. J. Minnaard, Chem. Commun., 2010, 46, 2535 RSC.
- (a) B. L. Feringa, B. D. Lange and J. C. de Jong, J. Org. Chem., 1989, 54, 2471 CrossRef CAS; (b) A. van Oeveren and B. L. Feringa, J. Org. Chem., 1996, 61, 2920 CrossRef CAS; (c) A. van Oeveren, J. F. G. A. Jansen and B. L. Feringa, J. Org. Chem., 1994, 59, 5999 CrossRef.
- B. ter Horst, B. L. Feringa and A. J. Minnaard, Org. Lett., 2007, 9, 3013 CrossRef CAS.
- B. M. Trost, J. Waser and A. Meyer, J. Am. Chem. Soc., 2007, 129, 14556 CrossRef CAS.
- G. Zhu and E. Negishi, Chem.–Eur. J., 2008, 14, 311 Search PubMed.
- O. Robles and F. E. McDonald, Org. Lett., 2009, 11, 5498 CrossRef CAS.
- G. E. Keck, C. E. Knutson and S. A. Wiles, Org. Lett., 2001, 3, 707 CrossRef CAS.
- S. F. Sabes, R. A. Urbanek and C. J. Forsyth, J. Am. Chem. Soc., 1998, 120, 2534 CrossRef CAS.
- J. A. Henderson, K. L. Jackson and A. J. Phillips, Org. Lett., 2007, 9, 5299 Search PubMed.
- A. Furstner and T. Nagano, J. Am. Chem. Soc., 2007, 129, 1906 CrossRef.
- J. M. Harris and G. A. O'Doherty, Org. Lett., 2000, 2, 2983 CrossRef CAS.
- S. C. Sinha and E. Keinan, J. Org. Chem., 1997, 62, 377 Search PubMed.
- R. P. van Summeren, B. L. Feringa and A. J. Minnaard, Org. Biomol. Chem., 2005, 3, 2524 RSC.
- M. F. Semmelhack and R. J. Hooley, Tetrahedron Lett., 2003, 44, 5737 CrossRef CAS.
- J. Thibonnet, V. Launay, M. Abarbri, A. Duchene and J. Parrain, Tetrahedron Lett., 1998, 39, 4277 CrossRef CAS.
- K. Ghosh, Y. Wang and J. T. Kim, J. Org. Chem., 2001, 66, 8973 CrossRef CAS.
- R. F. Newton and D. P. Reynolds, Tetrahedron Lett., 1979, 20, 3981 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: General procedures for the synthesis of substrates and products; 1H and 13C NMR spectral data of all products. See DOI: 10.1039/c1ob06700a |
| This journal is © The Royal Society of Chemistry 2012 |