A cationic surfactant assisted selective etching strategy to hollow mesoporous silica spheres†
Xiaoliang
Fang
,
Cheng
Chen
,
Zhaohui
Liu
,
Pengxin
Liu
and
Nanfeng
Zheng
*
State Key Laboratory for Physical Chemistry of Solid Surfaces and Department of Chemistry, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen, 361005, China. E-mail: nfzheng@xmu.edu.cn
First published on 8th February 2011
Abstract
Hollow mesoporous silica spheres have recently attracted increasing attention. However, effective synthesis of uniform hollow mesoporous spheres with controllable well-defined pore structures for fundamental research and practical applications has remained a significant challenge. In this work, a straightforward and effective “cationic surfactant assisted selective etching” synthetic strategy was developed for the preparation of high-quality hollow mesoporous silica spheres with either wormhole-like or oriented mesoporous shell. The as-prepared hollow mesoporous silica spheres have large surface area, high pore volume, and controllable structure parameters. Our experiments demonstrated that cationic surfactant plays critical roles in forming the hollow mesoporous structure. A formation mechanism involving the etching of solid SiO2 accelerated by cationic surfactant followed by the redeposition of dissolved silica species directed by cationic surfactant is proposed. Furthermore, the strategy can be extended as a general strategy to transform silica-coated composite materials into yolk-shell structures with either wormhole-like or oriented mesoporous shell.
Introduction
Design and synthesis of hollow micro-/nanostructures have been intensively pursued in the past decade.1–4 As a special type of these materials, hollow mesoporous silica spheres (HMSS) have attracted increasing attention especially in drug delivery and confined catalysis because of their several fascinating properties such as large surface area, excellent mechanical and thermal stability, low toxicity, high biocompatibility, high drug loading capacity, and highly permeable porous shell for easy loading and release of guest species.5–9 In previous studies, the most widely used strategies for synthesis of HMSS are hard-template10–16 and soft-template17–23 methods. The hard-template method was typically processed in multiple steps including the fabrication of sacrificial hard templates and their surface functionalization followed by the deposition of mesoporous silica shell and selective etching of the core templates. When uniform inorganic or polymer beads are used as sacrificial templates, although it is possible to obtain highly uniform HMSS, the required multi-step procedures are, in general, time-consuming and somewhat cumbersome. On the other hand, the soft-template method using micelle and vesicles reduces working procedure of the after-treatment, though the synthesis of the soft templates themselves may require complex procedures. Nevertheless, because of the difficulty in maintaining the uniformity of the soft templates in solution, the size distribution for HMSS made by the soft-template method is typically poor. Consequently, facile synthetic routes to high-quality HMSS are desirable for their practical applications.Very recently, new synthetic methods based on a silica-based etching strategy have been successfully developed to fabricate uniform HMSS and rattle-type mesoporous silica spheres. In these reported methods, solid silica spheres or modified solid silica spheres were used as the templates to create HMSS by selectively etching out the interior silica.9,24–29
As compared with other methods, such a selective etching strategy is facile and economical. Unfortunately, due to the lack of an effective way to control the pore formation, the pores in the HMSS particles obtained by the reported selective etching strategy were usually disordered and broadly distributed.9,24–29 However, for many applications of mesoporous silica materials, ordered and narrow-distributed pores are critical. For example, many recent reports have demonstrated that the chemical modification of the uniform pores in MCM-41 type materials can be used to regulate drug release and even fabricate controllable drug-release nanodevices.30–33 Therefore, from the viewpoint of both fundamental research and practical applications, there is a significant need to develop facile and effective strategies for preparing uniform HMSS with controllable well-defined pore structures.
In this study, we report a straightforward and effective “cationic surfactant assisted selective etching” (CSASE) route to high-quality HMSS with either wormhole-like or oriented mesoporous shell. Simply by treating solid SiO2 spheres with a Na2CO3 solution in the presence of a cationic surfactant (i.e.CTAB), HMSS particles with wormhole-like shell (denoted as HMSS-W) were easily prepared. By investigating the roles of CTAB, we proposed an etching-redeposition mechanism for the formation of HMSS-W from the solid SiO2 spheres and revealed the importance of CTAB in stabilizing CTAB/SiO2 mesostructures from alkaline etching. Such understanding was then used to prepare HMSS particles with oriented mesoporous shell (denoted as HMSS-O) by selectively etching the solid SiO2 core from the pre-made core-shell SiO2@CTAB/SiO2 spheres with oriented mesoporous structure. The pore channels in HMSS-O are aligned perpendicularly to the surface of spheres. By using CTAB as the template, the pores in both HMSS-W and HMSS-O are narrowly distributed around 2.5 nm. More importantly, the synthetic strategy developed in this work can be extended to fabricate yolk-shell nanostructures with functional nanoparticles encapsulated in HMSS, making the strategy useful and widely applicable for preparing advanced mesoporous materials for catalysis and drug delivery.
Experimental
Chemicals
Tetraethyl orthosilicate (TEOS) was purchased from Alfa Aesar. Ammonia solution (25–28%), cetyltrimethyl ammonium bromide (CTAB), cetyltrimethyl ammonium chloride (CTAC), ethanol, acetone, and sodium carbonate were purchased from Sinopharm Chemical Reagent Co. (Shanghai, China). Dodecyltrimethylammonium bromide (C12TAB) was purchased from TCI (Shanghai) Development Co., Ltd. All the reagents were used without further purification. Deionized water was used in all experiments.Synthesis of solid SiO2 spheres (denoted as sSiO2)
sSiO2 were synthesis following a modified Stöber method. In a typical synthesis, 6 mL of TEOS were rapidly added into a mixture of ethanol (74 mL), deionized water (10 mL), and ammonium aqueous solution (25–28%, 3.15 mL). The mixture was then stirred at room temperature for 1 h, resulting in the formation of a white silica colloidal suspension. The silica particles were centrifugally separated from the suspension and washed with deionized water and ethanol.Synthesis of HMSS-W
50 mg of the as-prepared sSiO2 were homogeneously dispersed in 9 mL of deionized water by ultrasonication for 15 min. After the addition of 1 mL CTAB aqueous solution (12.5 mg mL−1), the resultant mixture was further stirred at room temperature for 30 min before the introduction of Na2CO3 (212 mg). After the reaction was stirred at 35 °C for 24 h, the products were collected by centrifugation and washed with deionized water and ethanol.Synthesis of sSiO2@CTAB/SiO2
50 mg of the as-prepared sSiO2 was homogeneously dispersed in 10 mL of deionized water by ultrasonication for 15 min. The suspension was then added into a solution containing CTAB (75 mg), deionized water (15 mL), ethanol (15 mL), and ammonia solution (0.275 mL). After the mixture was stirred at room temperature for 0.5 h, 0.125 mL of TEOS was added quickly. After the reaction with stirring for 6 h, the products were collected by centrifugation, and redispersed in 10 mL of deionized water.Synthesis of HMSS-O
With vigorous stirring, 212 mg of Na2CO3 were added into the well-sonicated water suspension of the above sSiO2@CTAB/SiO2. After the reaction was stirred at 50 °C for 10 h, the products were collected by centrifugation and extensively washed with deionized water and ethanol.Removal of CTAB from the as-prepared products
Calcination or extraction was used to remove CTAB from the as-prepared products. For the calcination process, the as-prepared products were heated to 550 °C in air at a rate of 1.5 °C min−1 and maintained for 6 h. For the extraction process, the as-prepared products were dispersed in 80 mL of acetone and refluxed at 80 °C for 48 h. The extraction was repeated three times to fully remove CTAB. The final products were collected and washed with deionized water. After removal of CTAB, the sample sSiO2@CTAB/SiO2 was designated as sSiO2@mSiO2.Characterization
Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images were taken on a Hitachi S-4800 microscope with a field-emission electron gun and a TECNAI F-30 high-resolution transmission electron microscope operating at 300 kV, respectively. To prepare samples for SEM or TEM observations, one drop of ethanol suspension was deposited onto either silicon wafer (for SEM) or carbon-coated copper grid (for TEM). The structure of the products was also characterized by X-ray powder diffraction (XRD, Panalytical X'pert PRO diffractometer with Cu Kα radiation). The surface area was measured by the Brunauer–Emmett–Teller (BET) method using nitrogen adsorption and desorption isotherms on a Micrometrics ASAP 2020 system. The pore size distribution plot was obtained by the Barrett–Joyner–Halenda (BJH) method. The zeta potentials were measured on Nano-ZS (Malvern). The pH of the solutions was recorded with pH meter S20 (Mettler Toledo).Results and discussion
Facile conversion of sSiO2 into uniform HMSS-W
When simply treated by Na2CO3 in the presence of CTAB in an appropriate concentration, as illustrated in Fig. 1, the solid Stöber spheres were successfully converted into uniform hollow spheres (see Fig. S1 for the SEM images†). As revealed by the high-magnification TEM image (Fig. 1c), the shell of the as-obtained HMSS displays an obvious wormhole-like mesoporous structure. The mesostructure in HMSS-W was further confirmed by the powder X-ray diffraction (XRD). While the original sSiO2 does not diffract at low angles, the as-prepared HMSS-W exhibits a definite diffraction peak at 2.1° (2θ) with a weak and broad shoulder peak around 3.9°, which is typical of the wormhole mesostructures. The rather thin and curved feature of the mesoporous shell and therefore small diffraction domains might also account for the relatively broad primary peak and absence of well-defined high-order diffraction peaks.12 After thermal calcination to remove CTAB, the porosity of HMSS-W was investigated by N2adsorption-desorption measurements. As shown in Fig. 1f, the calcined HMSS-W exhibits a type IV isotherm with a type H2 hysteresis loop, characteristic of mesoporous materials. The BET surface area and pore volume of HMSS-W are 669.0 m2 g−1 and 0.97 cm3 g−1, respectively. In comparison, the BET surface area of sSiO2 is only 22.0 m2 g−1. As revealed by the pore size distribution of HMSS-W obtained from the analysis of the adsorption branch of the isotherm using the BJH method, HMSS-W has a rather narrow size distribution centered at 2.5 nm, similar to that of MCM-41. We thus believe that the pore formation in the HMSS-W is related to CTAB.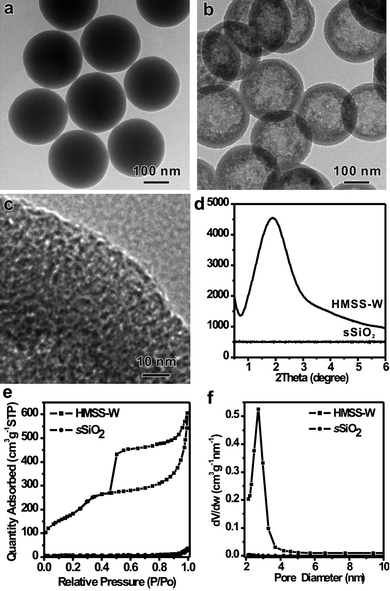 | ||
| Fig. 1 TEM images: (a) sSiO2, (b, c) HMSS-W, (d) low-angle X-ray diffraction pattern of sSiO2 and HMSS-W, and (e, f) N2 sorption isotherms and the pore size distribution of sSiO2 and HMSS-W. | ||
Roles of CTAB in the formation of HMSS-W
In order to understand the essential role of the CTAB in the formation of HMSS-W, a series of experiments were carried out by varying the CTAB concentration while keeping all other reaction conditions the same. Surprisingly, no hollow spheres were obtained in the absence of CTAB (Fig. 2a). Although the surface of sSiO2 became slightly rough, no significant size shrinking of sSiO2 was observed either, indicating that the etching process of sSiO2 caused by Na2CO3 alone was negligible. The alkaline etching of silica by Na2CO3 was considerably accelerated by the presence of CTAB. When CTAB was present in a concentration of 0.1 mg mL−1, sSiO2 were converted into core-shell spheres with a solid core and a thin layer of disordered mesoporous shell (Fig. 2b). The average diameter of the yolk was 200 nm, smaller than that of the unetched sSiO2 (245 nm). This size reduction gave evidence to the fact that CTAB promoted the etching of sSiO2 by Na2CO3. However, what was the reason behind such a promotion? The simultaneous formation of disordered mesoporous shell during the etching process provided us with some hints to solve this puzzle. At the CTAB concentration of 0.1 mg mL−1, the outer mesoporous shell was rather thin and even discontinuous. When the CTAB concentration was increased to 0.3 mg mL−1, a continuous mesoporous shell with an average thickness of 13 nm was completely formed on all particles while the core was further etched out (Fig. 2c). Careful observation also revealed that the more the yolk was etched, the thicker was the porous shell. When the CTAB concentration was further increased to 0.6 mg mL−1, the etching degree of the core was further extended and the thickness of mesoporous shell was increased to about 25 nm (Fig. 2d). If 1.25 mg mL−1CTAB was used, all solid spheres were eventually converted into HMSS with the core extensively etched out after the 24 h reaction. The thickness of the porous shell was measured to be 40 nm (Fig. 1). | ||
| Fig. 2 TEM images show the effect of CTAB concentration on the conversion of sSiO2 to HMSS. The CTAB concentration used were (a) 0 mg mL−1, (b) 0.1 mg mL−1, (c) 0.3 mg mL−1, and (d) 0.6 mg mL−1. All the other reaction conditions are the same: 50 mg sSiO2, 212 mg Na2CO3, 35 °C for 24 h. Shown in the inset are the corresponding TEM image with higher magnification. | ||
In the absence of CTAB, no obvious etching of the sSiO2 was observed. The etching process was significantly accelerated by the use of CTAB and a higher CTAB concentration yielded a thicker mesoporous shell before the depletion of the sSiO2. This observation clearly implies that the etching promotion effect by CTAB is essentially driven by the redeposition of dissolved silica species assisting by CTAB. Therefore, in the conversion process of sSiO2 to HMSS, CTAB should at least play the following two important roles: 1) the soft template to direct the formation of the mesoporous structure in the shell by co-assembly with the dissolved silica species; 2) the promoter to drive the dissolution dynamics of the sSiO2 and therefore accelerate their etching.
Formation mechanism of HMSS-W
Based on the above discussion over the roles of CTAB, we propose in Scheme 1 a formation mechanism of HMSS-W from sSiO2. Three important processes are involved in the conversion of sSiO2 into the hollow mesoporous structure: 1) adsorption of CTAB on the surface of sSiO2, 2) slow dissolution of SiO2 in the alkaline solution to form soluble silicate species, and 3) co-assembly of CTAB with soluble silicate species to form the mesostructure.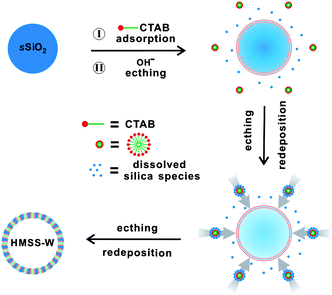 | ||
| Scheme 1 Schematic illustration for the formation of HMSS-W. | ||
Under the conditions of our procedure (35 °C, pH = 10.7), the dissolution of sSiO2 by Na2CO3 alone is very slow. However, at this pH, the sSiO2 spheres are negative charged, thereby benefiting the adsorption of cetyltrimethylammonium cations (CTA+) on their surface. As evidenced by zeta-potential measurements (Fig. 3), the adsorption of CTA+ makes the sSiO2 spheres positively charged with a zeta-potential of 4.0 mV. Since the silicate species etch-released from sSiO2 are anionic, the positive charge of the CTA+-adsorbing sSiO2 surface helps the re-deposition of the soluble silicate. Such a silicate redeposition process consumes some released silicate species and thus kinetically drives the dissolution reaction of sSiO2. More importantly, the redeposition of the etched-out silicate allows more adsorption of CTA+ (Fig. 3) and eventually leads to the formation of the mesostructured CTAB/SiO2 shell on the surface of sSiO2 spheres. Such an CTA+-promoted etching-redeposition formation mechanism is different from that involved in Yin's process.24,25
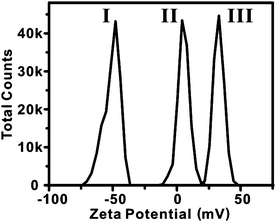 | ||
| Fig. 3 The distribution of zeta potentials of sSiO2 (I), CTAB-adsorbing sSiO2 (II), and HMSS-W (III). | ||
In order to demonstrate that soluble silicate species were indeed released from sSiO2, we designed an elaborate experiment to capture some of the dissolved silicate species by adding non-silica particles (i.e. rice-like α-Fe2O3) into the synthesis procedure (Fig. 4a). At the end of the reaction, the formation of core-shell structure with the core of the rice-like α-Fe2O3 particle can be observed by SEM (Fig. 4b) and TEM (Fig. 4c). The elemental mapping by EDX analysis (Fig. 4d-f) confirms the presence of a silica layer on each α-Fe2O3 particle, suggesting that the released silicate species can be easily re-condensed to form silica network. The proposed etching-redeposition mechanism was also supported by the size measurements on the as-prepared HMSS-W spheres. According to the mechanism, the initial co-assembly deposition of CTA+ with silicate released from the core occurred on the surface of the original sSiO2. Therefore, one would predict that the outer diameter of HMSS-W should be somewhat larger than the diameter of the original sSiO2. Indeed, starting from sSiO2, with an average diameter of 245 ± 10 nm (Fig. S2†), the outer diameter of the obtained HMSS-W was measured to be 260 ± 10 nm (Fig. S2†).
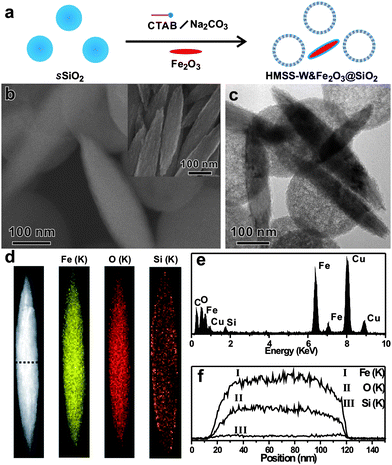 | ||
| Fig. 4 (a) Schematic illustration of capturing dissolved silicate species by rice-like α-Fe2O3 particles, (b, c) SEM and TEM images of as-obtained products (inset: SEM image of rice-like α-Fe2O3 particles), (d, e, and f) characterization of as-obtained single core-shell particle: (d) mapping of elements by EDX analysis under the STEM mode, (e) integrated EDX spectrum (the Cu signals are attributed to the TEM grid of copper used for the EDX experiments), (f) EDX line scanning profile. | ||
Considering that the essential role of CTAB during the formation of HMSS-W comes from the CTA+ cations, it was expected that the conversion of sSiO2 into HMSS-W could also be achieved by replacing CTAB with other quaternary cationic surfactants. Indeed, for instance, HMSS-W particles were also successfully prepared from sSiO2 by replacing CTAB with CTAC (Fig. S3†). When CTAB was substituted with C12TAB, hollow mesoporous spheres were successfully obtained. The pore size of the resultant HMSS was 2.1 nm, smaller than that of HMSS made by CTAB (Fig. S3†). By varying the size of cationic surfactants or using pore swelling agents, the CSASE strategy reported here should provide an effective method to create HMSS-W with tunable pore size over a wide range. Furthermore, the size of HMSS-W is also tunable by simply varying the size of the sSiO2 template. For instance, by the same surfactant-assisted etching procedures, the HMSS-W with particle sizes of 490 nm is obtained simply by using the sSiO2 with particle sizes of 470 nm (Fig. S4†).
Formation of HMSS with oriented mesoporous shell
For many applications, mesoporous materials with through pores running perpendicularly to their surface are highly desirable. Particularly for catalysis applications, such a pore structure would significantly shorten the diffusion length of target molecules and therefore enhance their overall catalytic performance. Unfortunately, the mesopores of the HMSS obtained by the one-step etching-redeposition strategy discussed above were wormhole-like. According to previous studies, the use of increased concentration of surfactant is important to obtain an ordered mesostructure rather than a wormhole-like mesostructure.36 We therefore increased the CTAB concentration to 10 mg mL−1 in our synthesis. However, no formation of hollow spheres with continuous mesoporous shell was observed. The reaction yielded smaller mesoporous silica particles surrounding the large SiO2 particles, which can be explained by the burst nucleation of a large number of small mesoporous silica particles on the surface of sSiO2 induced by the increased CTAB concentration (Fig. S5†). This result indicates the difficulty involved in the one-step conversion of sSiO2 into HMSS with mesopores oriented perpendicular to their surface.On the basis of the observation that the silicate-CTAB mesostructured shell of HMSS-W was fairly stable against the alkaline etching, we have switched to a two-step process to convert solid sSiO2 into HMSS with oriented pore structure. In this process, core-shell spheres sSiO2@CTAB/SiO2 with ordered mesoporous shell with perpendicular pores were first made. The solid SiO2 cores of the as-prepared sSiO2@CTAB/SiO2 were then selectively etched out by Na2CO3 to create the hollow spheres. In our experiments, the sSiO2@CTAB/SiO2 particles were prepared using modified literature methods.34,35 As clearly revealed by TEM (Fig. 5), the mesopores within the shell (about 50 nm in thickness) of the as-prepared sSiO2@CTAB/SiO2 were nicely oriented perpendicular to the surface of the spheres. As expected, after the alkaline treatment, the inner solid SiO2 core of each sSiO2@CTAB/SiO2 sphere was completely etched out while its outer shell remained intact, resulting in the formation of HMSS-O (see Fig. S6 for the SEM images†) with an oriented pore structure. As observed in the shell of the unetched sSiO2@CTAB/SiO2 spheres, all mesopore channels in HMSS-O were perpendicular to the surface of the spheres (indicated by arrows). After calcination, the BET surface area and pore volume of HMSS-O were measured to be 937 m2 g−1 and 0.77 cm3 g−1, respectively. In comparison, the calcined sSiO2@CTAB/SiO2 sample (sSiO2@mSiO2) had a BET surface area of 493 m2 g−1 and a pore volume of 0.41 cm3 g−1.
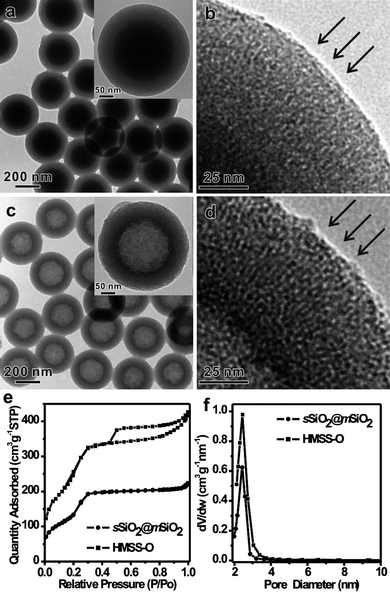 | ||
| Fig. 5 (a) TEM image of as-prepared sSiO2@CTAB/SiO2 (inset: typical TEM image of a single sSiO2@CTAB/SiO2 particle), (b) high-magnification TEM image corresponding to the dashed pane in inset of (a), (c) TEM image of as-prepared HMSS-O (inset: typical TEM image of single HMSS-O particle), (d) high-magnification TEM image corresponding to the dashed pane in inset of (c), and (e, f) N2 sorption isotherm and the pore size distribution of sSiO2@mSiO2 and HMSS-O. The orientation of the mesopores is indicated by arrows. | ||
The successful synthesis of HMSS-O from sSiO2@CTAB/SiO2 further confirms that, under the alkaline etching conditions, the CTAB/SiO2 mesostructure is more stable than the solid SiO2 core. Very recently, Shi's group also reported a better stability of mesostructured organic-containing silica shell made from TEOS and octadecyltrimethoxysilane.26 Based on 29Si MAS NMR studies, they attributed the better stability of the mesostructured shell to the higher degree of condensation in the mesoporous shell than in the solid core. But in our case of sSiO2@CTAB/SiO2, we hypothesized that the better stability of the CTAB/SiO2 mesostructure was mainly due to the presence of CTAB surfactant in the composite. Based on this hypothesis, a different result was expected if CTAB was removed from the CTAB/SiO2 mesostructured shell before alkaline etching. Therefore, in a control experiment, we removed CTAB extensively by acetone extraction. The obtained sSiO2@mSiO2 spheres were then subjected to alkaline etching by Na2CO3 at 50 °C. As shown in Fig. 6, no formation of HMSS was observed. After etching for 1 h, the mesoporous shell of sSiO2@mSiO2 spheres was heavily etched to form a disordered mesoporous shell but the solid core was only slightly etched (Fig. 6a). Upon increasing the etching time to 2 h, the mesoporous shell of sSiO2@mSiO2 spheres entirely disappeared and the diameter of the solid core decreased (Fig. 6b). This result reinforces our hypothesis that the presence of CTAB is the most important factor to stabilize the silicate-CTAB shell against alkaline etching.
 | ||
| Fig. 6 TEM image of sSiO2@mSiO2 etched by Na2CO3 for 1 h (a) and 2 h (b), inset: typical high-magnification TEM images. | ||
It is worth noting that the structure parameters of core-shell spheres sSiO2@CTAB/SiO2, such as the shell thickness and the core size, could be precisely tuned by varying the amount and the initial sizes of solid SiO2 core during the synthetic procedure (see Supporting Information†). As shown in Fig. 7a and b, by simply changing the initial amount of sSiO2, the shell thickness of core-shell spheres sSiO2@CTAB/SiO2 can be tuned to 100 nm. After the alkaline etching, the wall thickness of as-obtained HMSS-O is about 100 nm, meanwhile keeping the pore orientation perpendicular to the surface. HMSS-O with hollow interior sizes of 470 nm (Fig. 7c, d) and 130 nm (Fig. 7e and f) were obtained simply by using the sSiO2 with particle sizes of 470 nm and 130 nm as the templates for synthesis of sSiO2@CTAB/SiO2.
 | ||
| Fig. 7 TEM characterization of HMSS-O with different structure parameters: (a) HMSS-O with wall thickness of 100 nm, (b) high-magnification TEM image corresponding to the dashed pane in inset of (a), (c) HMSS-O with hollow interior size of 470 nm, (d) high-magnification TEM image corresponding to the dashed pane in inset of (c), (e) HMSS-O with hollow interior size of 130 nm, (f) high-magnification TEM image corresponding to the dashed pane in inset of (e). Oriented mesopores are indicated by arrows. | ||
Preparation of yolk-shell particles with wormhole-like/oriented mesoporous shell
Yolk-shell structures with individual nanocrystals encapsulated in hollow porous particles have been recently demonstrated not only to effectively enhance the stability of nanocrystals for catalysis applications, but also to introduce more functionality for bio-applications.37–39 Such yolk-shell structures were commonly fabricated by coating silica-encapsulated nanocrystals with a layer of porous material followed by selective etching of silica.8,9,26,37 As described above, the CSASE strategy that we have developed in this study has significantly simplified the direct conversion of sSiO2 into hollow mesoporous structures. We were therefore intrigued to explore the possibility of using such a simplified method for the preparation of yolk-shell particles with mesoporous shells. To further demonstrate the effectiveness of the developed CSASE strategy in converting non-spherical SiO2 particles, ellipsoid-shaped α-Fe2O3@SiO2 core-shell particles were prepared for the demonstration. As shown in Fig. 8a–c, after 24 h etching by Na2CO3 in the presence of CTAB at 35 °C, all of the α-Fe2O3@SiO2 core-shell particles were converted into elliptical yolk-shell particles with the ellipsoid-shaped α-Fe2O3 yolk nicely encapsulated inside. Similar to HMSS-W prepared from sSiO2 in one-step, the mesopores in the obtained elliptical SiO2 shell were wormhole-like. When the two-step protocol was used for transformation of the ellipsoid-shaped α-Fe2O3@SiO2, we obtained elliptical yolk-shell particles with ordered mesoporous shell with pores oriented perpendicular to their surface (Fig. 8d–f). Such an oriented porous structure is similar to that observed for HMSS-O prepared using the two-step method. Furthermore, when using Au@SiO2 core-shell nanospheres to replace sSiO2, Au@SiO2yolk-shell structures with either wormhole-like or oriented mesoporous shells can also be successfully obtained via the proposed CSASE strategy (Fig. S7†). Based on these results, we believe that the CSASE strategy reported here is applicable to the synthesis of yolk-shell nanostructures with diverse compositions, structures and functions. | ||
| Fig. 8 (a) TEM image of ellipsoid-shaped α-Fe2O3@SiO2, (b, c) TEM images of ellipsoid-shaped α-Fe2O3@HMSS-W, (d) TEM image of ellipsoid-shaped α-Fe2O3@SiO2@CTAB/SiO2, and (e, f) TEM images of ellipsoid-shaped α-Fe2O3@HMSS-O. Oriented mesopores are indicated by arrows. | ||
Conclusions
In summary, we have developed a facile route to fabricate hollow mesoporous silica with a wormhole-like shell based on a CSASE strategy. The use of cationic surfactant is critical to the formation of the hollow mesoporous structure from sSiO2. The surfactant plays the following three important roles in the reported solid to hollow transformation process: 1) a soft template to direct the formation of the mesoporous structure in the shell, 2) a promoter to accelerate etching of sSiO2 (i.e. hard-template), 3) a stabilizer to protect the silicate-CTAB shell from alkaline etching. On the basis of such understanding of roles of the cationic surfactant, we have successfully converted solid sSiO2 into hollow mesoporous silica with oriented mesoporous structure by selectively etching the pre-made core-shell sSiO2@CTAB/SiO2 composite with oriented mesoporous structure. Such a CSASE strategy is remarkably effective and reproducible, providing a facile route to tailor the structure parameters of as-obtained HMSS by selecting the appropriate cationic surfactant and also sSiO2 template. Furthermore, the strategy can be extended as a general strategy to transform silica-coated composite materials into yolk-shell structures with either wormhole-like or oriented mesoporous shell. We thus believe that the strategy reported herein will stimulate chemists to explore its further promising applications, such as confined catalysis and controlled release.Acknowledgements
We thank NSFC (21021061, 20925103, 20923004, 20871100), MOST of China (2009CB930703, 2011CB932403), the Fok Ying Tung Education Foundation (121011), NSF of Fujian for a Distinguished Young Investigator Grant (2009J06005) and the Key Scientific Project of Fujian Province (2009HZ0002-1).Notes and references
- F. Caruso, R. A. Caruso and H. Möhwald, Science, 1998, 282, 1111–1114 CrossRef CAS.
- Y. G. Sun, B. Mayers and Y. N. Xia, Adv. Mater., 2003, 15, 641–646 CrossRef CAS.
- C. S. Peyratout and L. Dähne, Angew. Chem., Int. Ed., 2004, 43, 3762–3783 CrossRef.
- X. W. Lou, L. A. Archer and Z. C. Yang, Adv. Mater., 2008, 20, 3987–4019 CrossRef CAS.
- L. Du, S. J. Liao, H. A. Khatib, J. F. Stoddart and J. I. Zink, J. Am. Chem. Soc., 2009, 131, 15136–15142 CrossRef CAS.
- Y. F. Zhu, T. Ikoma, N. Hanagata and S. Kaskel, Small, 2010, 6, 471–478 CrossRef CAS.
- Y. F. Zhu, J. L. Shi, W. H. Shen, H. R. Chen, X. P. Dong and M. L. Ruan, Nanotechnology, 2005, 16, 2633–2638 CrossRef CAS.
- Q. Zhang, T. R. Zhang, J. P. Ge and Y. D. Yin, Nano Lett., 2008, 8, 2867–2871 CrossRef CAS.
- J. Lee, J. C. Park, J. U. Bang and H. Song, Chem. Mater., 2008, 20, 5839–5844 CrossRef CAS.
- B. Tan and S. E. Rankin, Langmuir, 2005, 21, 8180–8187 CrossRef CAS.
- F. J. Suárez, M. Sevilla, S. Álvarez, T. Valdés-Solís and A. B. Fuertes, Chem. Mater., 2007, 19, 3096–3098 CrossRef CAS.
- H. Blas, M. Save, C. P.PasettoBoissière, C. Sanchez and B. Charleux, Langmuir, 2008, 24, 13132–13137 CrossRef CAS.
- W. R. Zhao, M. D. Lang, Y. S. Li, L. Li and J. L. Shi, J. Mater. Chem., 2009, 19, 2778–2783 RSC.
- Y. F. Zhu, E. Kockrick, T. Ikoma, N. Hanagata and S. Kaskel, Chem. Mater., 2009, 21, 2547–2553 CrossRef CAS.
- Y. Yamada, M. Mizutani, T. Nakamura and K. Yano, Chem. Mater., 2010, 22, 1695–1703 CrossRef CAS.
- G. G. Qi, Y. B. Wang, L. Estevez, A. K. Switzer, X. N. Duan, X. F. Yang and E. P. Giannelis, Chem. Mater., 2010, 22, 2693–2695 CrossRef CAS.
- C. E. Fowler, D. Khushalani and S. Mann, Chem. Commun., 2001, 2028–2029 RSC.
- J. G. Wang, Q. Xiao, H. J. Zhou, P. C. Sun, Z. Y. Yuan, B. H. Li, D. T. Ding, A. C. Shi and T. H. Chen, Adv. Mater., 2006, 18, 3284–3288 CrossRef CAS.
- M. H. Yu, H. N. Wang, X. F. Zhou, P. Yuan and C. Z. Yu, J. Am. Chem. Soc., 2007, 129, 14576–14577 CrossRef CAS.
- Z. G. Feng, Y. S. Li, D. C. Niu, L. Li, W. R. Zhao, H. R. Chen, L. Li, J. H. Gao, M. L. Ruan and J. L. Shi, Chem. Commun., 2008, 2629–2631 RSC.
- J. G. Wang, F. Li, H. J. Zhou, P. C. Sun, D. T. Ding and T. H. Chen, Chem. Mater., 2009, 21, 612–620 CrossRef CAS.
- J. Liu, S. B. Hartono, Y. G. Jin, Z. Li, G. Q. Lu and S. Z. Qiao, J. Mater. Chem., 2010, 20, 4595–4601 RSC.
- X. J. Wu and D. S. Xu, Adv. Mater., 2010, 22, 1516–1520 CrossRef CAS.
- T. R. Zhang, J. P. Ge, Y. X. Hu, Q. Zhang, S. Aloni and Y. D. Yin, Angew. Chem., Int. Ed., 2008, 47, 5806–5811 CrossRef CAS.
- T. R. Zhang, Q. Zhang, J. P. Ge, J. Goebl, M. W. Sun, Y. S. Yan, Y. S. Liu, C. L. Chang, J. H. Guo and Y. D. Yin, J. Phys. Chem. C, 2009, 113, 3168–3175 CrossRef CAS.
- Y. Chen, H. R. Chen, L. M. Guo, Q. J. He, F. Chen, J. Zhou, J. W. Feng and J. L. Shi, ACS Nano, 2010, 4, 529–539 CrossRef CAS.
- Q. Zhang, J. P. Ge, J. Goebl, Y. X. Hu, Z. D. Lu and Y. D. Yin, Nano Res., 2009, 2, 583–591 CrossRef CAS.
- M. Roca and A. J. Haes, J. Am. Chem. Soc., 2008, 130, 14273–14279 CrossRef CAS.
- M. Grzelczak, M. A. Correa-Duarte and L. M. Liz-Marzán, Small, 2006, 2, 1174–1177 CrossRef CAS.
- B. G. Trewyn, I. I. Slowing, S. Giri, H. T. Chen and V. S. Y. Lin, Acc. Chem. Res., 2007, 40, 846–853 CrossRef CAS.
- S. Angelos, E. Johansson, J. F. Stoddart and J. I. Zink, Adv. Funct. Mater., 2007, 17, 2261–2271 CrossRef CAS.
- I. I. Slowing, B. G. Trewyn, S. Giri and V. S. Y. Lin, Adv. Funct. Mater., 2007, 17, 1225–1236 CrossRef CAS.
- B. G. Trewyn, S. Giri, I. I. Slowing and V. S. Y. Lin, Chem. Commun., 2007, 3236–3245 RSC.
- S. B. Yoon, J. Y. Kim, J. H. Kim, Y. J. Park, K. R. Yoon, S. K. Park and J. S. Yu, J. Mater. Chem., 2007, 17, 1758–1761 RSC.
- Y. H. Deng, D. W. Qi, C. H. Deng, X. M. Zhang and D. Y. Zhao, J. Am. Chem. Soc., 2008, 130, 28–29 CrossRef CAS.
- N. Baccile, D. Grosso and C. Sanchez, J. Mater. Chem., 2003, 13, 3011–3016 RSC.
- S. H. Liu and M. Y. Han, Chem.–Asian J., 2010, 5, 36–45 CAS.
- A. Guerrero-Martínez, J. Pérez-Juste and L. M. Liz-Marzán, Adv. Mater., 2010, 22, 1182–1195 CrossRef CAS.
- J. Liu, S. Z. Qiao, S. B. Hartono and G. Q. Lu, Angew. Chem., Int. Ed., 2010, 49, 4981–4985 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures for sSiO2 with different sizes, α-Fe2O3@HMSS-W, α-Fe2O3@HMSS-O, Au@HMSS-W, and Au@HMSS-O; SEM images of HMSS-W; the histogram of size distribution of sSiO2 and HMSS-W; TEM images and the corresponding pore size distributions of HMSS-W obtained by using the CTAC/C12TAB; TEM images of HMSS-W obtained by using the sSiO2 with particle sizes of 470 nm; TEM images of the product obtained by using the 10 mg mL−1CTAB in the synthetic procedure of HMSS-W; SEM images of HMSS-O; TEM images of Au@SiO2, Au@HMSS-W, Au@SiO2@CTAB/SiO2, and Au@HMSS-O. See DOI: 10.1039/c0nr00893a |
| This journal is © The Royal Society of Chemistry 2011 |