Confirmation of the electrostatic self-assembly of nanodiamonds†
Lan-Yun
Chang
a,
Eiji
Ōsawa
b and
Amanda S.
Barnard
*c
aMonash Centre for Electron Microscopy and School of Chemistry, Monash University, Clayton, VIC, Australia
bNanoCarbon Research Institute, AREC, Faculty of Textile Science and Technology, Shinshu University, Ueda, Nagano, Japan
cCSIRO Materials Science and Engineering, Clayton, Australia. E-mail: amanda.barnard@csiro.au; Fax: +61 3 9545 2059; Tel: +61 3 9545 7840
First published on 24th January 2011
Abstract
A reliable explanation for the underlying mechanism responsible for the persistent aggregation and self-assembly of colloidal 5 nm diamond nanoparticles is critical to the development of nanodiamond-based technologies. Although a number of mechanisms have been proposed, validation has been hindered by the inherent difficulty associated with the identification and characterisation of the inter-particle interfaces. In this paper we present results of high resolution aberration corrected electron microscopy and complementary computer simulations to explicate the features involved, and confirm the electrostatic interaction mechanism as the most probable cause for the formation of agglutinates and agglomerates of primary particles.
Diamond nanoparticles are emerging as an important material for a variety of high performance technologies.1 In particular, their optical2 and chemical properties,3,4 coupled with their lack of cytotoxicity,5 make them a preferred candidate for biomedical applications.6,7 This includes drug delivery in the treatment of cancer,6 heart disease8 and in regenerative medicine.9 It also includes biolabelling10 based on their fluorescence in the visible range,2 owing to a number of photoactive point defects11 that exhibit a high quantum yield,12 superior photostability13 and structural stability.14 Nanodiamonds are also being tested as components in the next generation of ultrasensitive contrast agents for magnetic resonance imaging.15 In each case, however, the realisation of technologies based on their properties is contingent on our ability to control nanodiamonds on an individual and collective basis. The formation of robust porous microstructures is highly desirable for drug delivery, whereas effective biomarkers must be discrete and as small as possible.
For many years, detonation nanodiamond, also referred to as ultrananodiamonds (UND), at around ∼4.8 nm in size, appeared set to lead in this endeavour, but development was hindered by an inability to separate samples into individual single-digit nanoparticles.16 These ultra-small nanodiamonds exhibit a very strong tendency to aggregate, and form robust superstructures on the order of ∼100 nm in size.17 For many years it was speculated that the aggregation was due to van der Waals interactions, but this hypothesis was dismissed when it was found that the agglomerates strongly resisted dispersion.18 It was then argued that chemical bonds between the individual particles were responsible, but this suggestion was inconsistent with the XRD results showing that the diamond nanoparticles remain discrete. More recently, in attempts to understand the nature of agglomerates, researchers discovered that (given suitable conditions) diamond nanoparticles spontaneously self-assembled into ordered structures.19 Based on these observations the terms agglomerate and agglutinate were introduced to distinguish between (apparently) random collections of diamond nanoparticles and the ordered superstructures, but the underlying mechanism for the formation of agglutinates remained elusive.16
The mechanism responsible for the strong but long ranged interactions between colloidal diamond nanoparticles came in 2007,20 when it was shown that individual particles exhibit anisotropic facet-dependent variations in the surface electrostatic potential. The (100) facets exhibited a positive net charge, and the (111) facets were either found to be charge neutral or to exhibit a net negative charge, depending upon whether surface graphitization was efficient. Given that diamond nanoparticles are polyhedral; this gives rise to a multi-pole that determined how nanodiamonds interact with each other21 and with other molecules.22
It was then shown that, due to the potential energy difference between different crystallographic orientations, preferred particle–particle orientations existed. Two thermodynamically favourable inter-particle configurations were identified, with positive (100)+ facets interfacing with near-neutral (111)0 facets, or negative (111)− facets interfacing with near-neutral (111)0 facets. In both cases a neutral facet was an essential participant, and these interactions were termed coherent interfacial Coulombic interactions (CICIs).
Alternative configurations arising from random electrostatic interactions were termed incoherent interfacial Coulombic interactions (IICIs). These are both distinct from random weak interactions such as van der Waals forces that generally result in loosely aggregated powders. Depending upon whether CICIs or IICIs dominated, nanodiamonds were predicted to form agglutinates or agglomerates, as described in Table 1. Until now, however, this prediction has not been experimentally corroborated, partly due to the inherent difficulty associated with extracting quantitative or visual information on the relative amounts of IICI and CICI within a given sample, and partly due to the complexity of the reaction environment. There are a number of dependent physical variables influencing the mesostructure of samples during characterization, such as temperature and pressure induced surface restructuring, forced evaporation of the surrounding medium, space charging and primary particle mobility. Once the existence of CICI and IICI is confirmed, more detailed studies of how these mechanisms may be used to engineer diamond nanoparticles samples may begin.
| Name | Size/nm | Interaction mechanism | |||
|---|---|---|---|---|---|
| Type | Nature | Configuration | Terminology | ||
| a Coherent interfacial Coulombic interaction. b Incoherent interfacial Coulombic interaction. c van der Waals aggregation. | |||||
| Primary particle | 4.7 | — | — | — | Isolated |
| Agglutinate | ca. 60 | Interfacial | Electrostatic | Ordered | CICI a |
| Agglomerate | 100–200 | Interfacial | Electrostatic | Random | IICI b |
| Powder | >1000 | Intergranular | van der Waals | Random | VDWA c |
In order to definitely confirm the preferred orientations for the strong and long-ranged, nanodiamond–nanodiamond interactions, a number of observations are required. Firstly, that the facet–facet separation distance is on the order of 0.19 nm, as predicted by the simulations (and described in ref. 21). Secondly, that the particle–particle interfaces are dominated by negative/neutral or positive/neutral interactions as predicted by the computational study.21 If this is found to be the case, this will be evidenced by the orientation of interfacing particles, and it is expected that one would observe linear superstructures such as chains and loops, depending on the relative prevalence of the different types of preferred interactions.21
In an attempt to identify either (or both) of these signatures, we have undertaken high-resolution transmission electron microscopy (HRTEM) of colloidal diamond nanoparticles. The dispersed nanodiamond particles are now routinely prepared at a rate of about 2 kg month−1 by a six-step procedure in Ueda, Japan,3 in which the major step is beads-milling with 30 µm zirconia beads, as described in the ESI†.
Conventional HRTEM operating at medium voltage (100–300 kV) has been used in the past to characterise the structure of diamond nanoparticles.23,24 However, due to the weak scattering power of single particles, and the effect of delocalisation due to aberrations introduced by the objective lens, interpretation of such HRTEM images has been difficult. Here, we use aberration-corrected TEM operated at 80 kV to image the diamond nanoparticles, particularly in an attempt to identify their interfacial structures. The advantage of using such an instrumental setup is that the aberration-corrector greatly reduces the delocalisation in HRTEM images,24 and operating at 80 kV minimises the radiation damage to the sample, and increases the image contrast due to a stronger effective interaction at such accelerating voltage.25 Nevertheless, it is still expected that TEM specimen preparation and exposing the sample under electron beam during imaging may have an impact on the sample, for the reasons highlighted above.
Even with this state-of-the-art imaging instrumentation, characterisation of agglutinates and agglomerates is still challenging, due to the three-dimensional nature of the aggregated superstructures, the co-existence of CICI and IICI within the one sample, and the existence of more than one allotrope. In most cases, these particles are decorated with a partial coating of the fullerenic sp2carbon around the diamond-like sp3 core; a structure which is known as a bucky-diamond.26 This is not unexpected, and is generally consistent with previous experimental observations,23 computational simulations20 and theoretical predictions.27 After careful preparation and patient examination, we were able to achieve sufficient dispersion and identify numerous regions of the sample only one particle layer in thickness. Examples of these results are shown in Fig. 1, 2 and 3, and the details of the Fourier transform analysis are provided in the ESI†.
 | ||
| Fig. 1 The arrow indicates (111)|(111) interface between two 4 nm sized nanodiamonds. The orientation and the separation of the (111) lattice fringes are also shown. | ||
In Fig. 1 we see an interface between two 4 nm diamond nanoparticles, with the {111} lattice planes clearly visible. We have indexed this interface, based on the orientation of the two participating particles and the angle between the interface and the {111} planes, and confirm that it is (111)|(111), which is consistent with the prediction that an interface between (111)− facets and near-neutral (111)0 facets will be stable. We can also see that this interface contains a layer of fullerenic carbon (darker contrast at this imaging condition), which is also consistent with the prediction that only one surface (at the interface) would be graphitized and negatively charged. Unfortunately, the facet–facet separation distance cannot be identified unambiguously in the case, due to the off-zone axis orientations of the particles and slight delocalisation arising from the residual aberrations.
In Fig. 2 we see a loop of 4 nm diamond nanoparticles surrounding a 4–6 nm void. In this case, due to the three-dimensional nature of the superstructure, the precise locations of the interfaces are obscured, and characterization of the interfacial orientations is not possible. However, it was predicted that agglutinates would contain voids.21 The limitations inherent in CICI self-assembly manifest as a tiling problem, and to date no space filling two dimensional solutions have been identified that do not contain a relatively high fraction of voids. Recent results from nitrogen adsorption isotherms of the dried powder demonstrated the presence of nano-voids, with a characteristic size and volume which matched the observed quantity of the nanophase water identified using differential scanning calorimetry (DSC).28 The size of the voids identified during our study is consistent with these measurements28 and those predicted.21
 | ||
| Fig. 2 A three-dimensional (3-D) loop of nanodiamonds. The size of the individual nanodiamond ranges from 4–5 nm, and the size of the void within the loop is 4 nm (vertical width) by 6 nm (horizontal width). Due to the 3-D configuration, the locations of the interfaces are obscured. | ||
Fig. 3 presents a chain of four detonation nanodiamonds. The orientation of the nanodiamonds with respect to one another is determined by analysing the angles between the interface and the {111} lattice fringes, and the orientation of the particles as determined by the Fourier transforms of the sub-regions of individual nanodiamond. The location of three interfaces is highlighted by the arrows (marked a, b and c), and the fourth interface cannot be uniquely determined. We see here that the interfaces of these nanodiamonds are not of the (111)−|(111)0 type as shown in Fig. 1, but are a combination of {111} and {110} facets (formally {220}).
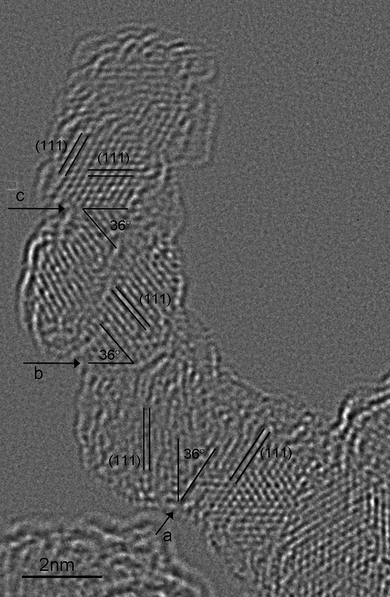 | ||
| Fig. 3 A linear chain of four ∼4.5 nm sized nanodiamonds. Their interfaces are determined to be (111)|(220) type (labeled as a, b and c), with one interface that cannot be uniquely determined. | ||
Interfaces containing {110} facets have not been considered before, and since a (111) facet is also involved, they may be negative, near-neutral or positive and still satisfy the selection rules (above). The surface electrostatic potential of (110) facet was briefly investigated in ref. 20. Although the results suggested that the facets were charge neutral on average, the cuboid morphologies were experimentally unrealistic, so the assignment of charge was inconclusive.
We have therefore undertaken density functional tight binding (DFTB) simulations on three rhombic dodecahedral nanodiamond structures, enclosed entirely by {110} facets, using the same computational methods as described in refs. 20 and 21. These results are presented in Fig. 4, where we can see that the surface facets are indeed near-neutral, with a slight positive charge. This suggests that (in order to obey the selection rules) the interfaces observed experimentally are due to the CICI mechanism, and involve negative charged (111)− facets that are fully graphitized, and near-neutral (110)0 facets.
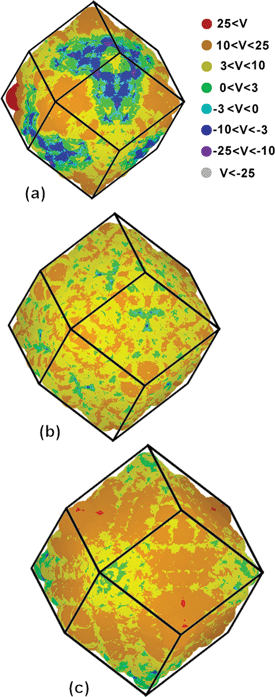 | ||
| Fig. 4 DFTB simulations of the surface electrostatic potential of dodecahedral diamond nanoparticles containing (a) 844 atoms, (b) 1232 atoms, and (c) 1722 atoms, measuring 2.2 nm, 2.5 nm and 2.9 nm, respectively. | ||
One issue that emerges from these observations and simulations is the lack of (100)+|(111)0 interfaces that were predicted to be almost as energetically favourable as the (111)−|(111)0 interactions. A more extensive search failed to find evidence of (100)+ facets interfacing with near-neutral (111)0 facets at all. To understand why this is the case we must first think about what is happening to the nanodiamonds during imaging. Although the graphitized surfaces are conducting, and will allow for the excess charge deposited by the electron beam to transport, the diamond cores are insulating, and many of the surfaces have not efficiently graphitized. This means that much of the charge deposited on the particles will continue to reside there, and will logically relocate to the (100)+ facets first, to balance the net positive potential and recover neutrality. Once all of the {100}+ sites have been satisfied, the incident electrons will begin to decorate the near-neutral (111)0 and (110)0 facets. Therefore, artificial anionic charging at doses consistent with imaging would disrupt the CICI mechanism, converting (100)+|(111)0 interfaces to (100)0|(111)0 interfaces is subject to weak van der Waals forces.
To test this hypothesis we have undertaken a series of DFTB simulations investigating the effect of excess charge on pairs of interacting diamond nanoparticles. Shown in Fig. 5a are the potential energy wells for (100)+|(111)0 and (111)−|(111)0 type interfaces at the lowest energy inter-facet angle of rotation. By taking the pairs of particles at the configuration corresponding to the minima of each well, and systematically injecting electrons (where Ne is the total number of electrons in the system), we can see the charge-dependent change in the well-depth in Fig. 5b. In the case of the (100)+|(111)0 interface, the interaction becomes rapidly more unstable with increasing anionic charge, and eventually becomes a forbidden interaction at 0.045 extra electrons per carbon atom (where Nc is the total number of carbon atoms in the system). Experimentally, an electron dose rate of approximately 2000 electrons per Å2 per second was used for HRTEM imaging. For an area where a single 4 nm single particle falls within the incident electron illumination, this dose rate is translated into approximately 600 electrons per atom per second. Assuming there is no charge dissipation, or transport to other diamond nanoparticles, a dose sufficient to separate a (100)+|(111)0 interface is achieved in approximately 75 microseconds, thereby explaining why this interface is not observed.
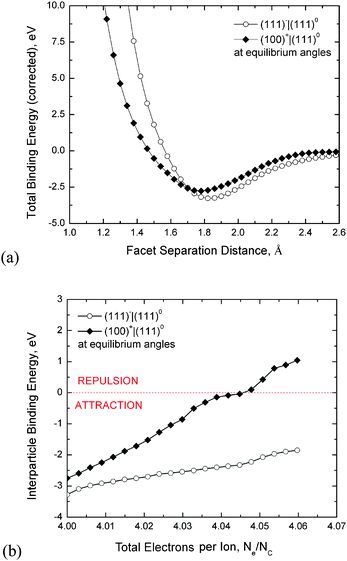 | ||
| Fig. 5 (a) The potential energy well due to CICI between nanodiamonds calculated using DFTB simulations, and (b) the effect of injected excess charge on the stability of (111)0|(111)− or (100)+|(111)0 interfaces; where Ne is the total number of electrons in the system, and Nc is the total number of carbon atoms. Increasing the number of excess electrons (anionic charges) over these pairs results in the satisfaction of the net positive potential on the (100)+ facets as the system recovers surface charge neutrality. This leads to weakening of the electrostatic (100)+|(111)0 interactions and eventually to separation of this nanodiamond pair. The term “equilibrium angle” refers to the lowest energy angle of rotation about the facet–facet normal. | ||
These results have another interesting consequence, being the prospect of controlling these interactions with electronic charge. This has been shown before using variations in pH,19 which is also important in drug delivery,7 but also introduces the opportunity to disperse the diamond nanoparticle sample without the need for mechanical milling with zirconia beads.18
The confirmation of the CICI interactions, and the possibility of controlling them using charge, also suggest that substrate patterning may be possible, which would be a great advance in the field of MEMS and NEMS.29 Currently diamond nanoparticles are an indispensable ingredient in the seeding of ultrananodiamond thin films,30 and it has already been demonstrated that self-assembled diamond nanoparticles may act as a template for subsequent film growthviachemical vapour deposition.31 Electrostatic dispersion and assembly of diamond nanoparticles on a substrate has already been demonstrated,32–34 and one can see why the tantalising possibility of controlled pre-patterning of substrate features on the order of 6–5 nm would be a revolution.
Therefore, future work will be directed toward processing methods aimed at exercising more control over the shape of individual nanodiamonds, to understanding how difference facet-dependent surface coatings and functionalization will affect the results, and whether these features can also be used to deliver greater specificity.
Acknowledgements
Computational resources for this project have been supplied by the National Computing Infrastructure (NCI) national facility under Partner Grant q27. Aberration-corrected Titan 80-300 (FEI Company) used in this project has been funded by Australian Research Council Grant LE0454166. E. O. is obliged to Bilateral International Collaborative R&D Program GT-2009-CL-OT-0058 administered by KIST.Notes and references
- Nanodiamonds: Applications in Biology and Nanoscale Medicine, ed. D. Ho, Springer Science + Business Media, Inc., Norwell, MA, USA, 2010 Search PubMed.
- J. Tisler, G. Balasubramanian, B. Naydenov, R. Kolesov, B. Grotz, R. Reuter, J.-P. Boudou, P. A. Curmi, M. Sennour, A. Thorel, M. Börsch, K. Aulenbacher, R. Erdmann, P. R. Hemmer, F. Jelezko and J. Wrachtrup, ACS Nano, 2009, 3, 1959 CrossRef CAS.
- E. Ōsawa, Chemistry of Single-Nano Diamond Particles, in Chemistry of Nanocarbons, ed. F. Wudl, S. Nagase and K. Akasaka, John Wiley & Sons, Oxford, UK, 2010, ch. 17 Search PubMed.
- L. Dai, Carbon Nanotechnology: Recent Developments in Chemistry, Physics, Materials Science and Device Applications, Elsevier, Amsterdam, 2006 Search PubMed.
- A. M. Schrand, J. Johnson, L. Dai, S. M. Hussain, J. J. Schlager, L. Zhu, Y. Hong and E. Ōsawa, Cytotoxicity and Genotoxicity of Carbon Nanomaterials, in Safety of Nanoparticles: from Manufacturing to Clinical Applications, ed. T. Webster, Springer Publishing, Brown University, Providence, RI, USA, 2008 Search PubMed.
- D. Ho, Therapy, 2009, 6, 99 CrossRef.
- A. M. Schrand, S. A. Ciftan Hens and O. A. Shenderova, Crit. Rev. Solid State Mater. Sci., 2009, 34, 18 Search PubMed.
- X.-Q. Zhang, M. Chen, R. Lam, X. Xu, E. Ōsawa and D. Ho, ACS Nano, 2009, 3, 2609 CrossRef CAS.
- R. A. Shimkunas, E. Robinson, R. Lam, S. Lu, X. Xu, X. Q. Zhang, H. Huang, E. Ōsawa and D. Ho, Biomaterials, 2009, 29, 5720 CrossRef.
- A. S. Barnard, Analyst, 2009, 134, 1751 RSC.
- Y. Y. Hui, Y.-R. Chang, T.-S. Lim, H.-Y. Lee, W. Fann and H.-C. Chang, Appl. Phys. Lett., 2009, 94, 013104 CrossRef.
- Y.-R. Chang, H.-Y. Lee, K. Chen, C.-C. Chang, D.-S. Tsai, C.-C. Fu, T.-S. Lim, Y.-K. Tzeng, C.-Y. Fang, C.-C. Han, H.-C. Chang and W. Fann, Nat. Nanotechnol., 2008, 3, 284 CrossRef CAS.
- C.-C. Fu, H.-Y. Lee, K. Chen, T.-S. Lim, H.-Y. Wu, P.-K. Lin, P.-K. Wei, P.-H. Tsao, H.-C. Chang and W. Fann, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 727 CrossRef CAS.
- C. Bradac, T. Gaebel, N. Naidoo, J. R. Rabeau and A. S. Barnard, Nano Lett., 2009, 9, 3555 CrossRef CAS.
- L. M. Manus, D. J. Mastarone, E. A. Waters, X.-Q. Zhang, E. A. Schultz-Sikma, K. W. MacRenaris, D. Ho and T. J. Meade, Nano Lett., 2010, 10, 484 CrossRef CAS.
- E. Ōsawa, Diamond Relat. Mater., 2007, 16, 2018 CrossRef CAS.
- A. Krüger, F. Kataoka, M. Ozawa, T. Fujino, Y. Suzuki, A. E. Aleksenskii, A. Y. Vul' and E. Ōsawa, Carbon, 2005, 43, 1722 CrossRef.
- E. Ōsawa, Pure Appl. Chem., 2008, 80, 1365 CrossRef CAS.
- H. Huang, L. Dai, D. H. Wang, L.-S. Tan and E. Ōsawa, J. Mater. Chem., 2008, 18, 1347 RSC.
- A. S. Barnard and M. Sternberg, J. Mater. Chem., 2007, 17, 4811 RSC.
- A. S. Barnard, J. Mater. Chem., 2008, 18, 4038 RSC.
- E. Ōsawa, D. Ho, H. Huang, M. V. Korobov and N. N. Rozhkova, Diamond Relat. Mater., 2009, 18, 904 CrossRef CAS.
- K. Iakoubovskii, K. Mitsuishi and K. Furuya, Nanotechnology, 2008, 19, 155705 CrossRef.
- P. W. Chen, Y. S. Ding, Q. Chen, F. L. Huang and S. R. Yun, Diamond Relat. Mater., 2000, 9, 1722 CrossRef CAS.
- E. J. Kirkland, Advanced Computing in Electron Microscopy, Plenum Press, New York, 1998 Search PubMed.
- J. Y. Raty, G. Galli, C. Bostedt, T. W. van Buuren and L. J. Terminello, Phys. Rev. Lett., 2003, 90, 037401 CrossRef.
- A. S. Barnard, S. P. Russo and I. K. Snook, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 73406 CrossRef.
- M. V. Korobov, M. M. Batuk, N. V. Avramenko, N. I. Ivanova, N. N. Rozhkova and E. Ōsawa, Diamond Relat. Mater, 2010, 19, 665 CrossRef CAS.
- O. A. Williams, M. Nesladek, M. Daenen, S. Michaelson, A. Hoffman, E. Ōsawa, K. Haenen and R. B. Jackman, Diamond Relat. Mater., 2008, 17, 1080 CrossRef CAS.
- O. A Williams, O. Douheret, A. Daenen, K. Haenen, E. Ōsawa and M. Takahashi, Chem. Phys. Lett., 2007, 445, 255 CrossRef CAS.
- M. L. Terranova, S. Orlanducci, E. Tamburri, V. Guglielmotti, F. Toschi, D. Hampai and M. Rossi, Nanotechnology, 2008, 19, 415601 CrossRef.
- J.-H. Kim, S.-K. Lee, O. M. Kwon and D.-S. Lim, J. Nanosci. Nanotechnol., 2009, 9, 4121 CrossRef CAS.
- J.-H. Kim, S.-K. Lee, O. M. Kwon, S. I. Hong and D.-S. Lim, Diamond Relat. Mater., 2009, 18, 1218 CrossRef CAS.
- S.-K. Lee, J.-H. Kim, M.-G. Jeong, M.-J. Song and D.-S. Lim, Nanotechnology, 2010, 21, 505302 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Details of the sample preparation and computational methodology. See DOI: 10.1039/c0nr00883d |
| This journal is © The Royal Society of Chemistry 2011 |