An enzyme-sensitive probe for photoacoustic imaging and fluorescence detection of protease activity†
Xiaohu
Xia
ab,
Miaoxin
Yang
a,
L. Kyle
Oetjen
a,
Yu
Zhang
a,
Qingge
Li
b,
Jingyi
Chen‡
*a and
Younan
Xia
*a
aDepartment of Biomedical Engineering, Washington University in St Louis, MO 63130, USA. E-mail: xia@biomed.wustl.edu; chenj@uark.edu; Tel: +1 314-935-8328
bEngineering Research Centre of Molecular Diagnostics, School of Life Sciences, Xiamen University, Xiamen, Fujian 361005, P. R. China
First published on 11th January 2011
Abstract
A gold nanocage and dye conjugate has been demonstrated for use with photoacoustic imaging and fluorescence detection of protease activity. The detection sensitivity could be maximized by using gold nanocages with a localized surface plasmon resonance peak away from the emission peak of the dye. These hybrids can be potentially used as multimodal contrast agents for molecular imaging.
Multimodal probes or contrast agents have garnered great interest in biomedical imaging over the past few years.1,2 Among them, inorganic nanoparticles have become versatile platforms due to their unique and often tunable physical/chemical properties that can enhance imaging contrast through different mechanisms.3 The surfaces of inorganic nanoparticles can also be readily conjugated with various functional groups for molecular imaging and targeted delivery. To this end, tremendous efforts have been directed towards quantum dots and iron oxide nanoparticles conjugated with functional moieties for detection by PET/optical or MRI/optical dual imaging modalities.4 There are only a few reports on the demonstration of multimodal probes based on Au nanoparticles and their conjugates. In one example, a 2.4-nm Au nanoparticle was conjugated with Gd-DTPA on its surface at a density of 50 Gd per Au nanoparticle and then used as a dual contrast agent for MRI/CT imaging.5 In another example, the oligonucleotide-conjugated Au nanoparticles were functionalized with Cy5.5 through the complementary oligonucleotides and then tested for fluorescence/reflectance imaging application.6 Here we report a new probe based on Au nanocages and fluorescence dyes coupled together through enzyme-cleavable peptides. This new probe can be potentially used as a dual contrast agent for photoacoustic/fluorescence imaging and therefore visualization of cellular distribution and function in living organisms.
In the previous studies, we have demonstrated Au nanocages — nanostructures with hollow interiors and porous walls — as a platform for various biomedical applications such as contrast enhancement in optical imaging, photothermal therapy, and controlled release.7 Owing to their tunable localized surface plasmon resonance (LSPR, including both absorption and scattering) features in the near-infrared (NIR) region and compact sizes (<50 nm), Au nanocages are good candidates as contrast agents for optical imaging. For example, they can strongly absorb light in the NIR region and thus drastically enhance the contrast for photoacoustic (PA) imaging. When they were intravenously injected into rats, the optical absorption in the cerebral cortex could be increased by up to 81%.8Gold nanocages have also been explored as tracers for non-invasive lymph node mapping with PA imaging at a depth of 33 mm below the skin surface, making them well-suited for humans.9 When conjugated with a targeting ligand, Au nanocages can triple the PA signals from a tumor as compared to nanocages covered with poly(ethylene glycol).10 In the present work, we attach fluorescent dyes to the surface of Au nanocagesviaprotease-cleavable peptides to construct a new probe that can be used for PA imaging and simultaneous detection of protease activity by fluorescence in the targeted lesion. In the initial constructs, the fluorescence emitted by the dye molecules is quenched due to energy transfer. However, in the presence of a protease, the dye molecules will be cleaved and released from the surface of the nanocage, and the fluorescence will be recovered. In practice, the distribution of Au nanocages can be mapped by PA imaging while the protease activity can be monitored by fluorescence spectroscopy or microscopy. We chose the matrix metalloproteases (MMPs) for our proof-of-concept experiments because they have long been recognized as a biomarker associated with cancer cell invasion and metastasis.11 For example, several clinical studies have shown that breast cancer patients have high levels of expression of MMPs, such as MMP-2, MMP-7, and MMP-9.12 More recent studies indicated that metastatic lymph nodes of breast cancer patients also overexpressed these enzymes.13 The probe demonstrated here can be potentially used as a new class of multimodal contrast agents for photoacoustic/fluorescence imaging in the diagnosis of cancer metastasis.
Fig. 1 shows a schematic illustration of the MMP-sensitive probe (dye–GKGPLGVRGC–Au nanocage) consisting of dye molecules and a Au nanocage coupled together through a peptide specific to MMP-2. Initially, the fluorescence emitted by the dye is quenched by the Au nanocage due to nano-surface energy transfer (NSET) mechanism. Similar to Förster resonance energy transfer (FRET), the excited state of the dye molecule (donor) can non-radiatively transfer its energy to a metal nanoparticle (acceptor) through the dye's dipole and the nearly free conduction electrons in the metal.14 Compared to FRET, the probability of the dipolar interactions for NSET is much greater because the conduction electrons in the metal nanoparticle can provide numerous dipole vectors on its surface to readily accept energy from the donor. Thus, the efficiency of NSET is much higher than FRET, typically close to 100% within the energy transfer distance. Additionally, the energy transfer distance in NSET is nearly twice as long as the typical Förster distance (<10 nm) in FRET because NSET has a 1/d4 dependence on distance as opposed to 1/d6 relationship for FRET.15 Previous studies have shown that if the donor (i.e., dye) and acceptor (i.e., Au nanoparticle) are connected through a single-stranded DNA (ssDNA) spacer, such a probe can be “turned on” by changing from a hairpin structure to a rod-like structure of ssDNA and used for the detection of DNA mismatches.16 For another system, the dyes leave the surface of Au nanoparticles after cleavage by an enzyme, and their fluorescence is recovered.17
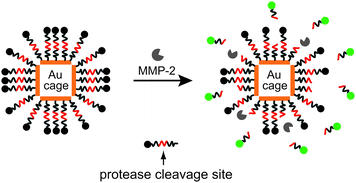 | ||
| Fig. 1 A schematic illustration of the dual probe that could be activated by an enzyme. The probe is comprised of a Au nanocage and fluorescent dyes linked together through an enzyme cleavable peptides. The emission from the dyes is initially quenched by the Au nanocage. Upon cleavage of the peptide and release of the dye from the surface of Au nanocage, the fluorescence of the dye is recovered. | ||
To fabricate the MMP-responsive probe, we conjugated a FITC-labeled C-terminus peptide (FITC–GKGPLGVRGC–NH2, GenScript, NJ) to the surfaces of Au nanocages (with an LSPR peak at 770 nm) through the free thiol group on the side chain of cysteine. The final product is denoted by FITC–GKGPLGVRGC–cage770. The sequence italicized in the peptide can be cleaved by MMP-2, a key protease expressed in tumors at much higher levels than in the healthy tissues. Briefly, 100 µL of 1 mM FITC-labeled peptide in DMF solution (0.1 µmol) was added to 1 mL of aqueous 1 nM cage770 solution (0.1 pmol) and incubated at room temperature for 24 h, followed by washing five times with 10 mM phosphate buffered saline (PBS, pH 7.4) to remove the excess peptide. To quantify the number of peptides conjugated to each nanocage, aqueous KCN solution at a final concentration of 100 mM was used to completely dissolve the Au nanocages, resulting in the release of all peptides from the nanocages into the surroundings. In Fig. 2a, the curves in green, red, and black represent fluorescence spectra taken from an aqueous suspension of cage770, and suspensions of the FITC–GKGPLGVRGC–cage770 (at a final concentration of 7 pM) before and after KCN etching, respectively. By comparing the fluorescence intensity with a calibration curve for the FITC-labeled peptide in Fig. S1†, we can estimate the number of peptides per nanocage and the NSET quenching efficiency. There were approximately 1.8 × 104peptides, on average, conjugated to the surface of each Au nanocage. We obtained a quenching efficiency of 96.6% by comparing fluorescence intensities before and after the FITC-labeled peptides had been released from the surfaces of the nanocages.
 | ||
| Fig. 2 (a) Fluorescence spectra of FITC–GKGPLGVRGC–cage770 before and after KCN etching; (b) fluorescence spectra of FITC–GKGPLGVRGC–cage770 before and after incubation with 1 U (72 ng mL−1) MMP-2 at 37 °C for 3 h. | ||
To demonstrate the ability to detect MMP-2 enzyme activity, we incubated the FITC–GKGPLGVRGC–cage770 conjugate with MMP-2 enzyme (an activated full-length, recombinant, human MMP-2 expressed by CHO cells, EMD Chemicals, NJ). Briefly, 2 mL of 10 pM FITC–GKGPLGVRGC–cage770 (20 fmol) in PBS was incubated with the active MMP-2 (1 U, 72 ng mL−1) at 37 °C for 3 h. Prior to enzyme cleavage, the fluorescence intensity of the solution was read as 18. After incubation with the MMP-2 enzyme for 3 h, the fluorescence dramatically increased to 69, about 4 times (∼280% increase in fluorescence intensity) stronger than what was observed before incubation with the enzyme (Fig. 2b).
We then examined the time dependence of enzyme activity using the same imaging probe. In this case, 20 fmol FITC–GKGPLGVRGC–cage770 in 2 mL PBS was incubated with the active MMP-2 (1 U, 72 ng mL−1) at 37 °C for different periods of time. The fluorescence intensity was linearly proportional to the incubation time in the period from 10 to 120 min and began to level off after 6 h (Fig. 3a). We also investigated the cleavage of FITC–GKGPLGVRGC–cage770 (20 fmol) with a fixed incubation time of 12 h but with different enzyme activities (Fig. 3b). The cleavage efficiency increased sharply below 1 U, but gradually leveled off. This result suggests that 1 U enzyme activity was sufficient to almost fully catalyze the cleavage of peptides in 20 fmol FITC–GKGPLGVRGC–cage770 during incubation. Fig. 3b also shows that 20 fmol of FITC–GKGPLGVRGC–cage770 probe can be used to detect the enzyme activity down to 0.01 U, corresponding to a sensitivity of 0.72 ng mL−1. To test the resistance of FITC–GKGPLGVRGC–cage770 probe against relevant inhibitors, we chose N-[(2R)-2-(hydroxamidocarbonylmethyl)-4-methylpentanoyl]-L-tryptophan (galardin), a potent cell-permeable MMP inhibitor with a molecular weight of 388.5, and performed dose-dependent experiments. We added various amounts of galardin up to 1 µM to the mixture of MMP-2 (1 U) and FITC–GKGPLGVRGC–cage770 (20 fmol), and then incubated for 2 h. Fig. 3c shows the dose–response study of galardin-mediated MMP inhibition. The inhibitory effect was approximately 72% when 1 µM of inhibitor was used. The Ki was estimated to be 300 pM, similar to the value reported in literature.18
 | ||
| Fig. 3 Characterization of the FITC–GKGPLGVRGC–cage770 probe: (a) percentage increase in fluorescence intensity when the FITC–GKGPLGVRGC–cage770 sample (20 fmol) was incubated with 1 U of MMP-2 as a function of time; (b) percentage increase in fluorescence intensity as a function of MMP-2 activity (20 fmol cage770–peptide–FITC, 12 h incubation); and (c) percentage increase in fluorescence intensity (1 U MMP-2, 20 fmol FITC–GKGPLGVRGC–cage770, 2 h incubation) as a function of MMP inhibitor concentration. The 50% inhibitory concentration in this assay was about 300 pM. Percentage increase in fluorescence intensity was defined as the change in fluorescence intensity divided by the initial fluorescence intensity. | ||
Finally, we embedded the mixture of FITC–GKGPLGVRGC–cage770 and MMP-2 in an agarose gel and performed a fluorescence imaging study. Briefly, 1 mL of FITC–GKGPLGVRGC–cage770 (20 pmol) was incubated with different doses of MMP-2 at 37 °C for 12 h. Then, 60 µL of the solution were mixed with 30 µL of 1.5% agarose, which was preheated at 90 °C for 1 h and immediately transferred into the wells of a 96-well plate. The mixture was allowed to cool to room temperature and solidified into gel within 15 min. We then took fluorescence micrographs using a QICAM Fast Cooled Mono 12-bit camera (Q Imaging, Burnaby, BC, Canada) attached to a fluorescence microscope (Olympus). All the images were captured with the same exposure parameters using Capture 2.90.1 and then converted into color images in Photoshop (Adobe). Fig. 4 shows fluorescence images of gel samples at various enzyme concentrations ranging from 0 to 10 U. The controls, plain agarose and agarose containing FITC–GKGPLGVRGC–cage770 with no MMP-2 displayed very similar, low fluorescence intensities. When 0.1 U (∼9 ng mL−1) of MMP-2 was applied to the sample, a slightly brighter color than those of the controls was observed, indicating that MMP-2 could be detected at a concentration of 9 ng mL−1. As the concentration of MMP-2 increased, the corresponding images became brighter. This result suggests the potential use of the dye–GKGPLGVRGC–Au nanocage probe in in vivo imaging of enzyme concentration or activity.
 | ||
| Fig. 4 Fluorescence images of FITC–GKGPLGVRGC–cage770 after incubation with different concentrations of MMP-2 at 37 °C for 12 h. The sample was mixed with 1.5% agarose to form gel for imaging. | ||
The NSET phenomenon always occurs when a dye molecule is in proximity to the metal nanoparticle regardless of the overlap between the emission spectrum of the dye and the LSPR peak of the nanoparticle. However, the fluorescence intensity of the free dyes strongly depends on the concentration of Au nanocages in the solution, as well as the spectral overlap between the fluorophore and the nanoparticle.19 We have studied different combinations of two fluorescent dyes (fluorescein isothiocyanate or FITC and Alexa Fluor® 790 or AF790) and 45-nm Au nanocages with LSPR peaked at two different wavelengths, 645 nm and 770 nm (cage645 and cage770, respectively). The results are shown in Fig. 5a and b. For FITC (ex. at 488 nm, and em. at 518 nm) at a concentration of 0.1 µM, the fluorescence intensity decreased as the concentration of Au nanocages increased (Fig. 5c). At a nanocage concentration of 10 pM, the fluorescence intensity of FITC dropped by 50% and 25% for cage645 and cage770, respectively, relative to the control sample without nanocages. A similar trend was also observed for AF790 (ex. at 780 nm and em. at 804 nm). At a nanocage concentration of 10 pM, the fluorescence intensity of AF790 (1 µM) decreased by 50% and 37.5% for cage770 and cage645, respectively, relative to the sample containing no nanocages. The decrease of fluorescence intensity in the presence of nanocages could be attributed to two factors: static quenching caused by diffusion of the free dyes to the surface of the nanocages and absorption of the nanocage at the emission wavelength of the dye.20 To maximize the intensity of emission from the free dye, the LSPR peak of the nanocages has to be tuned away from the emission peak of the dye. Hence, a combination of FITC–cage770 or AF790–cage645 would be an optimal choice for maximizing the detection sensitivity of the system. In addition, the concentration of nanocages should be kept below 10 pM to ensure a fluorescence recovery of >78% for FITC–cage770.
 | ||
| Fig. 5 Spectroscopic characterization of the Au nanocages and dyes: (a) excitation and emission spectra of FITC and extinction spectrum of Au nanocages with LSPR peaked at 770 nm (cage770); and (b) excitation and emission spectra of Alexa Fluor 790 (AF790) and extinction spectrum of Au nanocages with LSPR peaked at 645 nm (cage645). (c, d) Plots of the fluorescence intensity as a function of nanocage concentration where a specific amount of the dye was mixed with the Au nanocages of different concentrations in aqueous solutions: (c) FITC at a final concentration of 10−7 M and (d) AF790 at a final concentration of 10−6 M. | ||
In conclusion, we have demonstrated an enzyme-sensitive probe that is comprised of a Au nanocage and dye molecules linked together through an enzyme-cleavable peptide. By tuning the LSPR peak of the Au nanocage away from the emission peak of the dye, the fluorescence from the released dyes could be detected with relatively high sensitivity. The FITC–GKGPLGVRGC–cage770 probe was chosen as a model system for the detection of MMP-2. Both the fluorescence spectroscopy and microscopy studies show a high quenching efficiency for the dye and a high sensitivity in response to a relatively low concentration of enzyme and low enzyme activity. Due to the optical tunability of the nanocages in the NIR region, it is flexible to choose different dyes and nanocages to construct probes best-suited for in vivo imaging of cells and enzyme activities. This multimodal probe could enable cancer detection by photoacoustic imaging, cancer diagnosis by NIR fluorescence imaging, as well as cancer treatment by photothermal effect.
Acknowledgements
This work was supported in part by a Director's Pioneer Award from the NIH (DP1 OD000798), a grant from the NCI (R01 CA13852701), and a Research Development Award from the Alvin J. Siteman Cancer Center at Barnes-Jewish Hospital and Washington University School of Medicine. Siteman is supported by the Grant P30 CA91842 from the NIH. As a jointly supervised student, X.X. was also partially supported by a Fellowship from the China Scholarship Council. Part of the work was performed at the Nano Research Facility (NRF), a member of the National Nanotechnology Infrastructure Network (NNIN), which is supported by the NSF under award no. ECS-0335765.Notes and references
- A. Louie, Chem. Rev., 2010, 110, 3146 CrossRef CAS.
- (a) P. J. Ell, Br. J. Radiol., 2006, 79, 32 Search PubMed; (b) D. W. Townsend, T. Beyer and T. M. Blodgett, Semin. Nucl. Med., 2003, 33, 193 Search PubMed; (c) E. Tsukamoto and S. Ochi, Ann. Nucl. Med., 2006, 20, 255 Search PubMed.
- (a) J. H. Lee, Y. W. Jun, S. I. Yeon, J. S. Shin and J. Cheon, Angew. Chem., Int. Ed., 2006, 45, 8160 CrossRef; (b) W. J. Rieter, J. S. Kim, K. M. Taylor, H. An, W. Lin, T. Tarrant and W. Lin, Angew. Chem., Int. Ed., 2007, 46, 3680 CrossRef CAS; (c) P. Sharma, S. C. Brown, N. Bengtsson, Q. Zhang, G. A. Walter, S. R. Grobmyer, S. Santra, H. Jiang, E. W. Scott and B. M. Moudgil, Chem. Mater., 2008, 20, 6087 CrossRef CAS; (d) H. Xu, C. A. Regino, Y. Koyama, Y. Hama, A. J. Gunn, M. Bernardo, H. Kobayashi, P. L. Choyke and M. W. Brechbiel, Bioconjugate Chem., 2007, 18, 1474 CrossRef CAS; (e) C. A. Boswell, P. K. Eck, C. A. Regino, M. Bernardo, K. J. Wong, D. E. Milenic, P. L. Choyke and M. W. Brechbiel, Mol. Pharmacol., 2008, 5, 527 CAS; (f) S. Wang, B. R. Jarrett, S. M. Kauzlarich and A. Y. Louie, J. Am. Chem. Soc., 2007, 129, 3848 CrossRef CAS.
- (a) W. Cai, K. Chen, Z. B. Li, S. S. Gambhir and X. Chen, J. Nucl. Med., 2007, 48, 1862 CrossRef CAS; (b) M. L. Schipper, Z. Cheng, S. W. Lee, L. A. Bentolila, G. Iyer, J. Rao, X. Chen, A. M. Wu, S. Weiss and S. S. Gambhir, J. Nucl. Med., 2007, 48, 1511 CrossRef CAS; (c) J. H. Park, G. von Maltzahn, E. Ruoslahti, S. N. Bhatia and M. J. Sailor, Angew. Chem., Int. Ed., 2008, 47, 7284 CrossRef CAS; (d) R. Bakalova, Z. Zhelev, I. Aoki, H. Ohba, Y. Imai and I. Kanno, Anal. Chem., 2006, 78, 5925 CrossRef CAS; (e) S. S. Banerjee and D. H. Chen, Nanotechnology, 2009, 20, 185103 CrossRef; (f) J. Gunn, H. Wallen, O. Veiseh, C. Sun, C. Fang, J. Cao, C. Yee and M. Zhang, Small, 2008, 4, 712 CrossRef CAS; (g) M. J. Pittet, F. K. Swirski, F. Reynolds, L. Josephson and R. Weissleder, Nat. Protoc., 2006, 1, 73 CrossRef CAS.
- C. Alric, J. Taleb, G. Le Duc, C. Mandon, C. Billotey, A. Le Meur-Herland, T. Brochard, F. Vocanson, M. Janier, P. Perriat, S. Roux and O. Tillement, J. Am. Chem. Soc., 2008, 130, 5908 CrossRef CAS.
- N. Nitin, D. J. Javier and R. Richards-Kortum, Bioconjugate Chem., 2007, 18, 2090 CrossRef CAS.
- (a) S. E. Skrabalak, J. Chen, Y. Sun, X. Lu, L. Au, C. M. Cobley and Y. Xia, Acc. Chem. Res., 2008, 41, 1587 CrossRef CAS; (b) M. S. Yavuz, Y. Cheng, J. Chen, C. M. Cobley, Q. Zhang, M. Rycenga, J. Xie, C. Kim, K. H. Song, A. G. Schwartz, L. V. Wang and Y. Xia, Nat. Mater., 2009, 8, 935 CrossRef CAS; (c) J. Chen, C. Glaus, R. Laforest, Q. Zhang, M. Yang, M. Gidding, M. J. Welch and Y. Xia, Small, 2010, 6, 811 CrossRef CAS.
- X. Yang, S. E. Skrabalak, Z.-Y. Li, Y. Xia and L. V. Wang, Nano Lett., 2007, 7, 3798 CrossRef CAS.
- K. H. Song, C. Kim, C. M. Cobley, Y. Xia and L. V. Wang, Nano Lett., 2008, 9, 183.
- C. Kim, E. C. Cho, J. Chen, K. H. Song, L. Au, C. Favazza, Q. Zhang, C. M. Cobley, F. Gao, Y. Xia and L. V. Wang, ACS Nano, 2010, 4, 4559 CrossRef CAS.
- I. Stamenkovic, Semin. Cancer Biol., 2000, 10, 415 CrossRef CAS.
- J. E. Rundhaug, Clin. Cancer Res., 2003, 9, 551.
- A. Talvensaari-Mattila, P. Paakko and T. Turpeenniemi-Hujanen, Br. J. Cancer, 2003, 89, 1270 CrossRef CAS.
- S. H. Radwan and H. M. Azzazy, Expert Rev. Mol. Diagn., 2009, 9, 511 CrossRef CAS.
- (a) C. S. Yun, A. Javier, T. Jennings, M. Fisher, S. Hira, S. Peterson, B. Hopkins, N. O. Reich and G. F. Strouse, J. Am. Chem. Soc., 2005, 127, 3115 CrossRef CAS; (b) H. Szmacinski, K. Ray and J. R. Lakowicz, J. Biophotonics, 2009, 2, 243 CrossRef CAS.
- B. Dubertret, M. Calame and A. J. Libchaber, Nat. Biotechnol., 2001, 19, 365 CrossRef CAS.
- S. Lee, E. J. Cha, K. Park, S. Y. Lee, J. K. Hong, I. C. Sun, S. Kim, K. Choi, I. C. Kwon, K. Kim and C. H. Ahn, Angew. Chem., Int. Ed., 2008, 47, 2804 CrossRef CAS.
- D. R. Shalinsky, J. Brekken, H. Zou, C. D. McDermott, P. Forsyth, D. Edwards, S. Margosiak, S. Bender, G. Truitt, A. Wood, N. M. Varki and K. Appelt, Ann. N. Y. Acad. Sci., 1999, 878, 236 CrossRef CAS.
- M. P. Singh and G. F. Strouse, J. Am. Chem. Soc., 2010, 132, 9383 CrossRef CAS.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 1999 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Calibration curve showing fluorescence intensity as a function of the concentration of the FITC-labeled peptide in PBS (Fig. S1); TEM images of cage770 and cage645 (Fig. S2). See DOI: 10.1039/c0nr00874e |
| ‡ Current address: Department of Chemistry and Biochemistry, University of Arkansas, Fayetteville, AR 72701 USA. |
| This journal is © The Royal Society of Chemistry 2011 |