Probing the role of aromaticity in the design of dipeptide based nanostructures†
Aseem
Mishra
and
Virander Singh
Chauhan
*
International Centre for Genetic Engineering and Biotechnology, Aruna Asaf Ali Marg, New Delhi, 110067, India. E-mail: virander@icgeb.res.in
First published on 11th January 2011
Abstract
Self-assembly of peptide into nanostructures is believed to be stabilized primarily by aromatic interactions. Using a minimalistic approach, we probed the importance of aromatic interactions in the self-assembly of simple model dipeptides. Our results suggest that aromaticity may not be absolutely essential for self-assembly, even though it tends to provide directionality to the assembly. We found that peptides containing cyclic/linear side chain hydrophobic residues were also capable of forming stable self-assemblies that are stabilized by hydrophobic interactions. Our observations will find relevance in the design of small peptide based nanoparticles.
Introduction
Bottom-up fabrication of nanostructures by supramolecular assembly has long been considered as an efficient and inexpensive route for the manufacture of simple nanostructures.1 The feasibility of the process has been demonstrated repeatedly by the self-assembly of various organic structures ranging from porphyrins, to carbohydrates, nucleic acids, and proteins.1–4 Though, DNA and nucleic-acid based nano-structures have been of interest in the last few years, essentially because of the simplicity in the synthesis and a relatively good understanding of their folding rules, the development of peptide based nanostructures has amassed a greater interest in the design of complex nanostructures with functional significance.5,6 This is due to the inherent variety of structural designs offered by the building blocks along with the myriad of molecular interactions necessary for stabilizing the structures. Particularly interesting has been the demonstration of self-assembly in many designed dipeptides that has not only provided nanostructures with potential biomedical applications but has also served as a good platform for the investigation of structural features that influence the self-assembly of small peptide structures related to mis-folding related degenerative disorders (amyloids)7–9 as well as pre-biotic evolution of life.10,11Even though our understanding of molecular interactions dictating self-assembly of peptides is limited, aromatic (π-stacking) interactions seem to play a major role in their self-assembly. In fact, π-stacking interactions have always been the central theme in supramolecular chemistry because of their potential applications for molecular recognition, catalysis and structure stabilization.12–16 Although aromatic residues are important for the self-assembly of peptides, generic hydrophobic residues have also been shown to induce aggregation of amyloid derived peptides17,18 with the structures stabilized primarily by extensive and directional hydrogen bonding.
In this work, we investigated the essential role of aromaticity in small peptides in promoting self-assembly using a simple but well characterized dipeptide, Phe–Phe, that self-assembles into nanotubes.8 We systematically substituted the aromatic residues in Phe–Phe by a structurally similar residue, cyclohexylalanine (Cha), which instead has a six membered aliphatic ring. Self-assembly of synthetic model dipeptides, Cha–Phe, Phe–Cha and Cha–Cha, was investigated by transmission electron microscopy (TEM), circular dichroism (CD) spectroscopy and dynamic light scattering (DLS) techniques. We also analysed the self-assembly of other homo-aliphatic dipeptides containing naturally occurring hydrophobic amino acidsi.e.Val, Leu and Ile. Our results indicate that while aromatic residues may have inherent potential to facilitate and stabilize self-assembled structures; their presence is not a necessary requirement for self-assembly in small peptides. Our observations reflect on the possible relevance of aliphatic residues in the design of peptide based nano-structures as well gain a better understanding of amyloid formation.
Results and discussion
Self-assembly of the aromatic dipeptide Phe–Phe has been described previously and therefore we thought it to be a good template to address the role of aromaticity in the self-assembly of small peptides. However, we decided to characterize the self-assembly behaviour of Phe–Phe in greater detail so that a direct comparison could be made with the corresponding non-aromatic dipeptides (ESI, Table S1†). To assemble the peptide, a hexafluoroisopropanol (HFIP) stock solution (20 mg ml−1) of Phe–Phe was diluted in de-ionized double distilled water to a final peptide concentration of 1 mg ml−1, followed by removal of residual HFIP by vacuum evaporation. The peptide self-assembled into a mesh/network of long tube like structures with bulges along their length (even when imaged within the first hour of the sample preparation). The average diameter of the nanotubes was 45 nm (Fig. 1) and length in microns.8 The tubes exhibited an inherent tendency to form visibly larger clusters on standing for a few hours (1–2 h) at room temperature probably due to the increase in the network size with time. Dynamic light scattering studies on the sample showed a highly heterogeneous population of structures with an average hydrodynamic radius above 1000 nm suggestive of very large order assemblies/network size; the tubes did not appear isolated in any of the preparations. These observations were consistent with previous reports.8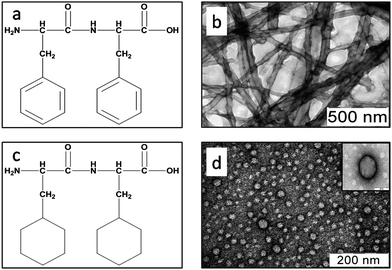 | ||
| Fig. 1 Chemical structures of (a) Phe–Phe and (c) Cha–Cha. Transmission electron micrographs of (b) Phe–Phe and (d) Cha–Cha, at a concentration of 1 mg ml−1 stained with 1% uranyl acetate. | ||
To probe the deterministic role of aromatic interactions in self-assembly, Phe residues in Phe–Phe were systematically substituted with Cha, the saturated analogue of Phe. Self-assembly properties of the dipeptides Cha–Phe and Phe–Cha were then analysed under the same conditions as for Phe–Phe. The electron micrographs showed that Cha–Phe assembled into a mixture of vesicular (average diameter ≈ 50 nm) and fibrillar assemblies (average diameter ≈ 20 nm). The fibrils appeared twisted along their length (Fig. 2a). Phe–Cha, on the other hand, assembled exclusively into pleomorphic vesicular structures with diameters ranging between 10 and 80 nm; no fibril formation was observed even upon ageing for 7 days (Fig. 2b). Interestingly, sequence shuffle has been demonstrated to influence the morphological consequence in self-assembly of tetra-peptides derived from antamanide.19 The shuffling of two amino acids Phe and Pro in the model peptide Phe-Phe-Pro-Pro that assembled primarily into nanoscopic ring like structures to form tubular (for Pro-Pro-Phe-Phe) and vesicular (for Pro-Phe-Phe-Pro) nanostructures suggested dissimilar aggregating pathways for the sequence shuffled peptides.19 In the case of non-aromatic dipeptide Cha–Cha, the electron micrographs showed well-defined vesicular structures with diameters ranging between 40 and 60 nm (Fig. 1). However, upon ageing the sample for over an hour at room temperature, visible aggregates could be seen in the sample resulting from the physical clustering of the smaller vesicles. However, unlike Phe–Phe, Cha–Cha did not assemble into fibrils/nanotubes even at higher concentrations (∼10 mg ml−1) or upon ageing (>14 days).20 These results suggested that while aromatic interactions may not be absolute pre-requirements for assembly of small peptides, the occurrence and positioning of aromatic residues can modulate features of the assembled structure. In fact, large stretches of aliphatic residue containing peptides have been shown to self-assemble into various geometries21,22 without the aid of aromatic interactions. Also, many different surfactants self-assemble by shear hydrophobic interactions. Our results indicate that a similar assembling behaviour can occur even in peptides as small as dipeptides.
 | ||
| Fig. 2 Transmission electron micrographs of (a) Cha–Phe and (b) Phe–Cha, at a concentration of 1 mg ml−1 stained with 1% uranyl acetate. | ||
We next studied the molecular structure of the assemblies formed by Cha–Cha to be able to compare it with the model aromatic peptide Phe–Phe. The CD spectrum of Phe–Phe nanotubes was characterized by two positive bands at 220 nm and 200 nm consistent with previous reports (Fig. 3a).23 However, Phe–Phe at the same concentration but in the non-assembled form (dissolved in HFIP) exhibited only the 220 nm band with a lower ellipticity, suggesting the uniqueness of the 200 nm positive band to the fibrillar form (Fig. 3a). Though, the 200 nm band has been attributed to π–π* transition and the 220 nm band to the n–π* transition of the peptide bond in the dipeptides, the exact origin of the CD bands is largely unknown. We speculate that the 220 nm band arises from the peptide backbone with the value of ellipticity indicative of the relative degree of hydrogen bonding while the lower wavelength 200 nm band appears to be contributed by the aromatic stacking in the fibrillar/nanotubular assembly. It is noteworthy that the lower wavelength band has also been observed in other self-assembling aromatic residue containing dipeptides (previous report24,25 and unpublished results from our lab) while it is absent in non-aromatic based assemblies (as in Cha–Cha and Ile–Ile (ESI S1†)). HD exchange 2D-COS-FTIR24,26 showed that the amide I peak occurring at 1620 cm−1 (Fig. 3c) is suggestive of the extended and directional H-bonding network of the peptide backbone in the self-assembled Phe–Phe, similar to β-sheet based amyloid fibres.8 Previous studies have also demonstrated that in solid-state Phe–Phe adopts an extended head-to-tail hydrogen bonding involving the free N- and C-terminus.27 Further, Phe–Phe assemblies also stained positively with ThT (ESI S2†), a widely used amyloid sensitive dye, ref. 28 and references therein, suggesting the occurrence of directional hydrogen bonded networks in the nanostructure.
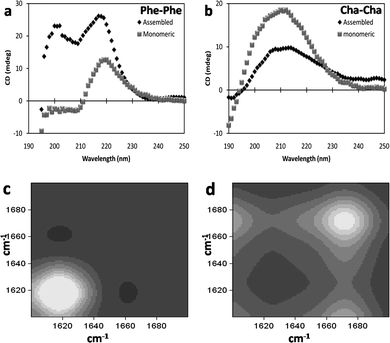 | ||
| Fig. 3 Far-UV circular dichroism spectra of (a) Phe–Phe and (b) Cha–Cha in assembled and monomeric forms. 2D-COS-FTIR spectra of amide I band of (c) Phe–Phe and (d) Cha–Cha assemblies indicating backbone conformation. | ||
The dipeptide Cha–Cha exhibited a broad positive CD band centering at 210 nm (Fig. 3b) in the assembled as well as the non-assembled form, though ellipticity decreased after its assembly into nano-vesicles. This observation was intriguing as it is generally accepted that a high degree of ordered H-bond networks occurs in polymer based self-assembled structures. We believe that the decrease in ellipticity in Cha–Cha nano-vesicles was indicative of non-structured intermolecular hydrogen bonds in the structure. The amide I peak at 1675 cm−1 in HD exchange 2D-COS-FTIR (Fig. 3d) of the nanovesicles was also suggestive of weak and non-structured hydrogen bonds. The kinetics of HD exchange revealed that the amide I band of Cha–Cha exchanged rapidly (within 6 min) compared to Phe–Phe suggestive of high solvent accessibility of the peptide bond (ESI S3†). Indirectly, it also suggested that the nanovesicles formed by Cha–Cha were porous allowing solvent molecules to penetrate into the structure;29 a feature that might have potential applications in the development of drug-delivery systems. HD exchange NMR of Cha–Cha in monomeric and assembled form also indicated that the peptide backbone was involved in hydrogen bonding (slow exchange) only to a small degree while the terminal amine was free (rapid deuterium exchange occurred in less than 2 min); however, aggregation resulted in significant dampening of the signal to make any substantial interpretations about the molecular structure. From the above studies, it appeared that the self-assembly of Cha–Cha was stabilized primarily by hydrophobic interactions of the side-chain with minor contribution of the peptide backbone hydrogen bonding. As expected, Cha–Cha nanovesicles did not cause enhancement of fluorescence upon binding to thioflavin T indicating the lack of extensive hydrogen bonding in the self-assembled structure.
Since, many hydrophobic aliphatic side-chain containing dipeptides have also been demonstrated to organize into higher order supramolecular assemblies in crystalline form, ref. 21 and references therein, and having shown that Cha–Cha self-assembles readily, we decided to investigate the self-assembly of naturally occurring linear side-chain containing dipeptides. To do this, three dipeptides Val–Val, Leu–Leu and Ile–Ile were synthesized and their assembly behaviour was studied under exactly the same conditions as described earlier. TEM showed that all the three homo-aliphatic dipeptides could assemble into vesicular structures (Fig. 4) with diameters ranging between 70 and 110 nm (Fig. 4). DLS studies also supported the results of electron microscopy even though the average hydrodynamic radius of Ile–Ile appeared to be marginally smaller (∼70 nm) than the Leu–Leu and Val–Val assemblies (∼100 nm) in solution (ESI S4†). However, none of the peptides formed fibrils or higher order structures upon ageing the preparations for two weeks or more. As expected, diglycine and dialanine were highly water soluble (due to their low hydropathy index) and did not exhibit any self-assembly.

To assess the stability of Cha–Cha nanovesicles in comparison to Phe–Phe nanotubes, we characterized the thermal stability of the assemblies by DLS wherein the scattering intensity and hydrodynamic radius of the assemblies were measured as a function of temperature. However, the inhomogeneous nature of the suspension and the clustering tendency of the Phe–Phe and Cha–Cha hindered this study. In order to obtain a homogenous preparation, the assemblies were sonicated for 10 min at 180 W (1 s: ON and 3 s: OFF pulse sequence) followed by cooling to room temperature. Consistent with the variable temperature CD experiment, both the assemblies exhibited an overall decrease in scattering intensity with increasing temperature suggesting melting of the assembly (Fig. 5). The melting temperature for the Phe–Phe nanotubes was 63 °C beyond which there was no further decrease in the scattering intensity (Fig. 3a). At this temperature and above, DLS revealed that the assembly dissociated to smaller oligomeric form whose hydrodynamic radius was less than 5 nm (below the detection limit of DLS setup used) (Fig. 3b). The CD spectrum of Phe–Phe at 80 °C exhibited a residual 200 nm band (with low ellipticity) (ESI S5†) indicating the occurrence of non-monomeric forms of the dipeptide even at high temperatures. Self-assembled Cha–Cha exhibited a melting temperature of 55 °C (Fig. 3a). Interestingly, beyond this temperature a much larger residual structure with a hydrodynamic radius of 138 nm was observed that was stable up to 95 °C (Fig. 5b). This was interesting as energetics of aromatic–aromatic interactions have been found to be more favourable than Phe–Cha or Cha–Cha pair.30–32 However, hydrophobic forces appear to dominate in the assembled Cha–Cha and provide thermodynamic stability. We also assessed the stability of Cha–Cha assemblies under different pH (=2, 7 and 11) conditions and found that they were largely unaffected. It seemed that pH affected only the physical clustering of the nano-vesicles as evidenced by the difference in their cluster size; low pH resulted in increased cluster size compared to neutral and alkaline pH (ESI S6†). These results suggest that changes in the ionization state of solvent exposed amine and carboxyl groups of the peptide probably aided cluster stabilization of Cha–Cha nanovesicles through electrostatic interactions. As expected, the nanovesicles also resisted proteolytic degradation (ESI S7†) by Proteinase K and trypsin in contrast to Phe–Phe assemblies.8 These studies imply that Cha–Cha nanostructures were stable and could therefore be developed further as drug delivery vehicles.
Conclusion
In conclusion, using a well-characterized aromatic dipeptides template, Phe–Phe, we have attempted to delineate the role of aromatic residues in the self-assembly of dipeptides. By systematic replacement with cyclohexylalanine and other non-aromatic residue containing dipeptides we have found that while aromatic residues favour fibrillar and/or tubular structures, they are not crucially required for self-assemblyper se. These findings may be useful for further design of novel self-assembling peptide systems using a judicious mix of aromatic and non-aromatic residues. It would be of interest to investigate the assembly behaviour of other homo- and hetero-aliphatic dipeptides to gain a definitive understanding of molecular determinants of peptide self-assembly. Studies are underway to explore the possibility of using the described system as a potential delivery agent.Materials and methods
Peptide synthesis
The relevant N-terminal protected amino acid (Boc-Xaa-COOH) (5 mM) was dissolved in dry tetrahydrofuran (Sigma-Aldrich) and the resulting solution stirred in an ice-salt bath at −15 °C. N-Methylmorpholine (Sigma) (0.65 ml, 5 mM) was added to the solution followed by isobutyl chloroformate (Sigma) (0.7 ml, 5 mM). After 30 min, a pre-cooled aqueous solution of the incoming amino acid (H-Yaa-COOH) (5.5 mM) and sodium hydroxide (0.22 g, 5.5 mM) was added and mixture stirred overnight at room temperature. The reaction mixture was concentrated in vacuo, acidified with citric acid to pH 3.0 and extracted with ethyl acetate (Spectrochem) (3 × 20 ml). The ethyl acetate layer was washed with water (2 × 15 ml), with saturated sodium chloride (1 × 20 ml), dried over anhydrous sodium sulfate and evaporated to yield Boc-Xaa-Yaa-COOH dipeptide. Deprotection at the α-amino group and side chain protection were achieved by treatment with 98% formic acid (30 ml) for 3 h or 50% TFA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) : DCM for 30 min at room temperature. The reaction mixture was evaporated to dryness and the residue was precipitated with anhydrous diethyl ether in cold (Cha–Cha dipeptide was precipitated with ice-cold double distilled water). The resulting precipitate was filtered, washed several times with dry ether and subsequently lyophilized from 10% acetic acid–water (20 ml) to yield the final compound. Synthetic yields were typically between 90 and 95%.
: DCM for 30 min at room temperature. The reaction mixture was evaporated to dryness and the residue was precipitated with anhydrous diethyl ether in cold (Cha–Cha dipeptide was precipitated with ice-cold double distilled water). The resulting precipitate was filtered, washed several times with dry ether and subsequently lyophilized from 10% acetic acid–water (20 ml) to yield the final compound. Synthetic yields were typically between 90 and 95%.
The peptides were purified on a preparative reverse phase C18 column (Deltapak, C18, 15µ, I.D. 300 × 19 mm) using acetonitrile–water linear gradient 5–45% acetonitrile (0.1%TFA)/water (0.1% TFA) at a flow rate of 4 ml min−1 over 25 min. The purified peptides were reinjected into an analytical reverse phase C18 column (Phenomenex, C18, 5µ, I.D. 250 × 4.6 mm) using a acetonitrile–water linear gradient 5–95% acetonitrile (0.1%TFA)/water (0.1% TFA) at a flow rate of 1 ml min−1 over 25 min. The purified peptides were verified by mass spectroscopy (Applied Biosystems QStar (Q-TOF)).
Self-assembly was initiated by dissolving 1 mg of a purified peptide in 50 µl of hexafluoroisopropanol (HFIP) (Sigma) and by subsequent addition of 1 ml of double distilled water. Samples were aged for 2–24 h prior to their characterization by TEM, CD and DLS.
Circular dichroism spectroscopy
All spectra were recorded on a JASCO-810 polarimeter equipped with a Peltier type thermostat and purged continuously with dry N2 gas at 10 l min−1 (LPM) during data acquisition. Data were collected in a quartz cuvette with a path-length of 0.1 cm or 1 cm between 190 nm and 250 nm at a scan speed of 25 nm min−1 and a response time of 16 s. The average of 50 scans was used for analysis of the spectrum.Fourier transform infrared (FTIR) spectroscopy
Spectra were collected on a Perkin-Elmer Spectrum BX FTIR spectrometer. The assembled peptide samples were spotted on a CaF2 window and air-dried at 99% relative humidity. The samples were then rehydrated with D2O, and hydrogen–deuterium exchange was monitored every minute for 10 min in the spectral range of 1600–1700 cm−1. The FTIR spectra were smoothed with a smoothing length of 20 units. Two-dimensional correlation spectroscopy (2D COS) maps were generated from the stack of spectra using the freely available software package 2D-Shige. All spectral assignments were done according to published reports.Electron microscopy
Peptide samples were adsorbed on a 300 mesh copper grid with formvar support and stained with 1% uranyl acetate and viewed under a 120 kV mode of a TEM (Tecnai 12 BioTWIN, FEI Netherlands). Photomicrographs were digitally recorded using a Megaview III (SIS, Germany) digital camera.Dynamic light scattering (DLS)
Light scattering studies were performed in Photocor complex. Dynamic light scattering experiment was carried out at 37 °C and an angle of 90° using 632 nm laser and Photocor-FC digital correlator operating in multiple-tau mode. The correlation curves were fitted using DYNAL-S (intensity distribution analysis) for the estimation of hydrodynamic radius. For measuring the thermal stability of the assemblies, the samples were prepared at the peptide concentration of 1 mg ml−1, vigorously sonicated and were subsequently diluted to an effective peptide concentration of 0.18 mg ml−1 at which the suspension was stable for the duration of data collection. Temperature dependent scattering intensity and hydrodynamic radius were measured and plotted.Nuclear magnetic resonance (NMR)
Cha–Cha was dissolved in d6-DMSO to a concentration of 6 mg ml−1. The monomeric nature of the preparation was ascertained using DLS. To assemble the peptide, D2O was added to a final concentration of 20%. Self-assembly of Cha–Cha under these conditions was again ascertained using DLS. All NMR experiments were carried out at 298 K on a Bruker Avance III spectrometer equipped with 5 mm cryogenic triple-resonance probes, operating at a field strength of 500 MHz. One-dimensional 1H NMR spectra were measured with 32 scans with a relaxation delay of 2 s. Free induction decays (FIDs) were collected with a spectral width of 8012.82 Hz and an acquisition time of 2.045 s (t1max). All NMR spectra were processed using Topspin 2.1 (Bruker AG).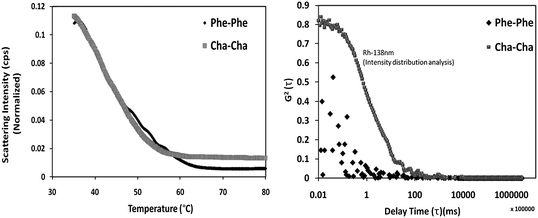 | ||
| Fig. 5 (a) Melting curves of Phe–Phe and Cha–Cha assemblies. (b) Correlation curves of the residual structure of the assemblies at 80 °C. Cha–Cha reveals a thermo-stable intermediate with Rh of 138 nm. | ||
Acknowledgements
The authors thank Ranjan Nanda and Rashmi Srivastav for mass spectrometry analysis. Authors also thank Neel Sarovar Bhavesh for help in obtaining and analysis of NMR spectra. Intellectual inputs from Jiban Jyoti Panda, Madhvi Gupta and Akash Saini during the course of this investigation are also acknowledged. The authors also thank Dr Atanu Basu, NIV, Pune for his support during TEM analysis. Financial support was received from ICGEB core grant and Wellcome Trust-DBT Alliance fellowship. Financial support for the NMR facility at the ICGEB, New Delhi was provided by the Department of Biotechnology, Government of India.References
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418–2421 CrossRef CAS.
- S. M. Douglas, H. Dietz, T. Liedl, B. Hogberg, F. Graf and W. M. Shih, Nature, 2009, 459, 414–418 CrossRef CAS.
- J. A. A. W. Elemans, R. van Hameren, R. J. M. Nolte and A. E. Rowan, Adv. Mater., 2006, 18, 1251–1266 CrossRef CAS.
- S. Zhang, Nat. Biotechnol., 2003, 21, 1171–1178 CrossRef CAS.
- G. Colombo, P. Soto and E. Gazit, Trends Biotechnol., 2007, 25, 211–218 CrossRef CAS.
- X. Zhao, F. Pan and J. R. Lu, Prog. Nat. Sci., 2008, 18, 653–660 CrossRef CAS.
- C. Wu, H. Lei and Y. Duan, Biophys. J., 2005, 88, 2897–2906 CrossRef CAS.
- M. Reches and E. Gazit, Science, 2003, 300, 625–627 CrossRef CAS.
- S. L. Gras, Aust. J. Chem., 2007, 60, 333 CrossRef CAS.
- I. Ghosh and J. Chmielewski, Curr. Opin. Chem. Biol., 2004, 8, 640–644 CrossRef CAS.
- M. Fishkis, Origins Life Evol. Biosphere, 2007, 37, 537–553 CrossRef CAS.
- G. Bellesia and J.-E. Shea, Biophys. J., 2009, 96, 875–886 CrossRef CAS.
- C. G. Claessens and J. F. Stoddart, J. Phys. Org. Chem., 1997, 10, 254–272 CrossRef CAS.
- E. Gazit, FASEB J., 2002, 16, 77–83 CrossRef CAS.
- G. B. McGaughey, M. Gagné and A. K. Rappé, J. Biol. Chem., 1998, 273, 15458–15463 CrossRef CAS.
- A. S. Shetty, J. Zhang and J. S. Moore, J. Am. Chem. Soc., 1996, 118, 1019–1027 CrossRef CAS.
- W. Kim and M. H. Hecht, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15824–15829 CrossRef CAS.
- C. Wurth, N. K. Guimard and M. H. Hecht, J. Mol. Biol., 2002, 319, 1279–1290 CrossRef CAS.
- K. B. Joshi and S. Verma, J. Pept. Sci., 2008, 14, 118–126 CrossRef CAS.
- J. Naskar and A. Banerjee, Chem.–Asian J., 2009, 4, 1817 CrossRef CAS.
- H. Xu, J. Wang, S. Han, J. Wang, D. Yu, H. Zhang, D. Xia, X. Zhao, T. A. Waigh and J. R. Lu, Langmuir, 2008, 25, 4115–4123.
- S. Santoso, W. Hwang, H. Hartman and S. Zhang, Nano Lett., 2002, 2, 687–691 CrossRef CAS.
- L. Adler-Abramovich, M. Reches, V. L. Sedman, S. Allen, S. J. B. Tendler and E. Gazit, Langmuir, 2006, 22, 1313–1320 CrossRef CAS.
- A. Mishra, J. J. Panda, A. Basu and V. S. Chauhan, Langmuir, 2008, 24, 4571–4576 CrossRef CAS.
- J. J. Panda, A. Mishra, A. Basu and V. S. Chauhan, Biomacromolecules, 2008, 9, 2244–2250 CrossRef CAS.
- I. Noda, Anal. Sci., 2007, 23, 139–146 CrossRef.
- C. Görbitz, Acta Crystallogr., Sect. B: Struct. Sci., 2010, 66, 84–93 CrossRef.
- A. A. Maskevich, V. I. Stsiapura, V. A. Kuzmitsky, I. M. Kuznetsova, O. I. Povarova, V. N. Uversky and K. K. Turoverov, J. Proteome Res., 2007, 6, 1392–1401 CrossRef CAS.
- M. Gupta, A. Bagaria, A. Mishra, P. Mathur, A. Basu, S. Ramakumar and V. Chauhan, Adv. Mater., 2007, 19, 858–861 CrossRef CAS.
- M. S. Searle and B. Ciani, Curr. Opin. Struct. Biol., 2004, 14, 458–464 CrossRef CAS.
- C. D. Tatko and M. L. Waters, J. Am. Chem. Soc., 2002, 124, 9372–9373 CrossRef CAS.
- D. M. Chung, Y. Dou, P. Baldi and J. S. Nowick, J. Am. Chem. Soc., 2005, 127, 9998–9999 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of experimental observations. See DOI: 10.1039/c0nr00691b |
| This journal is © The Royal Society of Chemistry 2011 |