Nanostructured nanoparticles of self-assembled lipid pro-drugs as a route to improved chemotherapeutic agents
Sharon M.
Sagnella†
a,
Xiaojuan
Gong†
ab,
Minoo J.
Moghaddam
a,
Charlotte E.
Conn
c,
Kathleen
Kimpton
d,
Lynne J.
Waddington
e,
Irena
Krodkiewska
c and
Calum J.
Drummond
*c
aCSIRO Materials Science and Engineering, PO Box 184, North Ryde, NSW 1670, Australia
bUniversity of Sydney, School of Chemistry, NSW 2006, Australia
cCSIRO Materials Science and Engineering, Private Bag 33, Clayton South MDC, VIC 3169, Australia. E-mail: calum.drummond@csiro.au
dCSIRO Food and Nutritional Sciences, PO Box 52, North Ryde, NSW 1670, Australia
eCSIRO Materials Science and Engineering, 343 Royal Parade, Parkville, VIC 3052, Australia
First published on 21st December 2010
Abstract
We demonstrate that oral delivery of self-assembled nanostructured nanoparticles consisting of 5-fluorouracil (5-FU) lipid prodrugs results in a highly effective, target-activated, chemotherapeutic agent, and offers significantly enhanced efficacy over a commercially available alternative that does not self-assemble. The lipid prodrug nanoparticles have been found to significantly slow the growth of a highly aggressive mouse 4T1 breast tumour, and essentially halt the growth of a human MDA-MB-231 breast tumour in mouse xenografts. Systemic toxicity is avoided as prodrug activation requires a three-step, enzymatic conversion to 5-FU, with the third step occurring preferentially at the tumour site. Additionally, differences in the lipid prodrug chemical structure and internal nanostructure of the nanoparticle dictate the enzymatic conversion rate and can be used to control sustained release profiles. Thus, we have developed novel oral nanomedicines that combine sustained release properties with target-selective activation.
Nanomedicine has emerged as a strategy to circumvent the high systemic toxicity associated with many current cancer treatments by allowing the sustained delivery of high drug payloads directly to the tumour site.1–8Prodrugs, a group of bioreversible derivatives of drug molecules that undergo transformation to an active drug through enzymatic and/or chemical transformation in vivo, are a well established strategy for providing improved tumor specificity.3–8 Likewise, dispersions of nanoparticulate self-assembled amphiphile systems, which have a high internal surface area, can be used to deliver both hydrophilic and hydrophobic payloads. Additionally, they possess an internal nanostructure that can dictate drug release rates,1,2,9–17 and are attractive candidates for providing sustained delivery of pharmaceutical agents.14,18 These two strategies have been fused creating, for the first time, prodrug analogues of the established chemotherapy agent 5-FU, which self-assemble into nanostructured systems producing a target-activated, sustained release system that can be delivered orally.
We focus herein on the design and development of self-assembled amphiphile molecules; prodrug analogues of 5-fluorouracil (5-FU), their higher order nanoassemblies, the in situ enzymatic hydrolyses and controlled release of the drug from different types of nanoassemblies, and the in vivo efficacy in breast cancer models in mice. 5-FU is one of the oldest chemotherapeutic agents and has played a dominant role for decades in the treatment of breast cancer and a variety of other solid tumours. A series of new prodrug amphiphile conjugates based on a commercially available 5-FU prodrug analogue, Capecitabine (N4-pentyloxycarbonyl-5′-deoxy-5-fluorocytidine, Xeloda®), have been developed by substituting the pentyl group at the N4 position with three different hydrophobic alkyl chains: palmityl (5-FCPal), oleyl (5-FCOle), and phytanyl (5-FCPhy) (Fig. 1A). Similar to Capecitabine, the hydrophobic chains were attached to the precursor 5-FC at the 4-N position via a carbamate bond, and thus these bonds are expected to be hydrolyzed by the same enzymatic route as Capecitabine. Following oral administration, the prodrug is expected to enter the blood stream through the gastrointestinal tract with its molecular structure intact, and is converted to 5-FU by multistep enzymatic reactions in the liver and at the tumor site in a similar manner to Capecitabine (Fig. 1B).4,19–21 The multistep prodrug bioconversion process provides targeted activation by constraining conversion of the 5-FC prodrug amphiphile conjugates specifically to the tumor site.
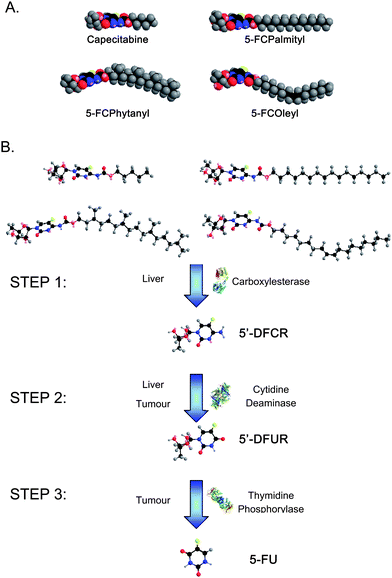 | ||
| Fig. 1 (A) Energy minimized space filling molecular models of Capecitabine, 5-FCPalmityl, 5-FCOleyl, and 5-FCPhytanyl. The unsaturated oleyl chain and branched phytanyl chain occupy a much larger effective chain volume than their straight chain counterparts, thus allowing them to self-assemble into inverse lyotropic liquid crystalline phases. (B) Schematic of the multi-step enzymatic conversion of 5-FC prodrugs to 5-FU in vivo. After oral administration, the prodrugs enter the blood stream through the gastrointestinal tract. Step 1 occurs mainly in the liver viacarboxylesterase cleavage of the prodrug to 5′-deoxy-5-fluorocytidine (5′-DFCR). Step 2 can occur in both the liver and at the tumour site where the enzyme cytidine deaminase is present and cleaves 5′-DFCR to 5′-deoxy-5-fluorouridine (5′-DFUR). Step 3, which results in the final cleavage of 5′-DFUR to 5-fluorouracil (5-FU) viathymidine phosphorylase occurs preferentially at the tumour site as the enzyme is over expressed in a large number of solid tumours. | ||
By using evolving amphiphile self-assembly materials by design rules22 the ability to control the internal nanostructure of nanoparticle dispersions of 5-FC analogues by alterations to the hydrophobic alkyl chain structure has been demonstrated. 5-FCPal was dispersed in excess water to form solid lipid nanoparticles with a crystalline lamellar lattice parameter of 28.7 Å (Fig. 2A). Introduction of an unsaturated hydrocarbon chain (5-FCOle) resulted in the formation of liposomes at room temperature and cubosomes at 37 °C.23 The cubosomes prepared for the in vivo studies consisted of co-existing gyroid (Ia3d symmetry: lattice parameter of 148.7 Å) and double-diamond (Pn3m symmetry: lattice parameter of 80.9 Å) architectures (Fig. 2B). Dispersions of 5-FCPhy, which contains an isoprenoid-type branched chain, contained a mixture of cubosomes again of gyroid (124.4 Å) and double-diamond (78.5 Å) architectures (Fig. 2C). The average size of these nanostructured nanoparticle dispersions, determined viadynamic light scattering, was dependent on the processing conditions and the hierarchical structure of the nanoassemblies; 5-FCPal (700 nm), 5-FCOle (255 nm) and 5-FCPhy (164 nm), which is consistent with cryo-TEM results.
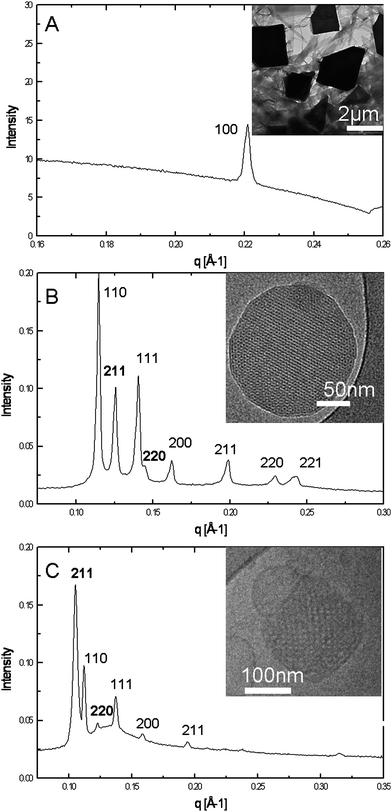 | ||
| Fig. 2 1-D diffraction patterns for (A) solid–lipid nanoparticles of 5-FCPal, (B) cubosomes of 5-FCOle, and (C) cubosomes of 5-FCPhy. In all cases the inset shows a representative cryo-TEM image from the same system. | ||
It is well established that the bioconversion rates of prodrugs can be adjusted through modifications to the molecular structure;5,6 however, in this case we have aimed to control drug release rates not only via changes to the molecular structure but also through differences in the internal nanostructure of the self-assembled prodrugs. Differences in drug release rates from the different prodrug nanoassemblies were assessed using carboxylesterase (CES) hydrolysis of the nanoparticle dispersions. The catalytic activity of CES is crucial for predicting the bioconversion rates of the prodrugs, and thus plays an important role in the onset of the drug conversion, the duration of release, and the intensity of the drug activity. Fig. 3 shows the CES hydrolysis rate for ∼1 mM of the three 5-FC lipid prodrugs and Capecitabine at 37 °C. At a CES enzyme concentration of 0.5 mg ml−1 (a CES concentration similar to that purified from human liver cytosol24), Capecitabine had fully hydrolyzed within 15 minutes, whereas full hydrolysis of the three self-assembled lipid prodrugs varied depending on the structure of the hydrocarbon chain. 5-FCOle, which forms cubic nanoparticles, displayed the fastest hydrolysis, with full hydrolysis occurring within ∼13 h. The cubic nanoassemblies formed by 5-FCPhy displayed a slightly longer full hydrolysis time of ∼26 h, whereas the 5-FCPal solid–lipid nanoparticles required more than a week for full hydrolysis to occur. As enzyme specificity is determined by the structure of the substrate active site, the observed differences in hydrolysis rates can be attributed to molecular and internal particulate nanostructure variations. The lack of self-assembly structure for Capecitabine and associated ease of access for the enzyme dictate a fast rate of hydrolysis. In contrast, the close crystalline packing and low fluidity of the 5-FCPal solid–lipid nanoparticles greatly hinders enzyme activity, resulting in an extremely slow rate of hydrolysis. The continuous water channels present in the cubic nanoparticles formed by 5-FCOle and 5-FCPhy facilitates ease of access to the substrate and a shortened hydrolysis rate. An acceleration of the hydrolysis rate observed for both the 5-FCOle and 5-FCPhy nanoparticle dispersions (occurring at around 10 h for the 5-FCOle and 20 h for the 5-FCPhy dispersions) is indicative of partial or complete breakdown in the internal nanostructure of the particle. As both 5-FCOle and 5-FCPhy possess similar internal cubic nanostructures, the difference in hydrolysis rate between these two nano-assemblies can be attributed to the molecular structure of their alkyl component. The branched phytanyl chain of 5-FCPhy sterically hinders enzymatic access to the cleavage site to a much greater degree than the singly unsaturated oleyl chain, with a resultant decrease in hydrolysis rate. Thus, alterations to both the molecular structure and the internal nanostructure of these nano-assemblies provide a simple method for controlling the release profile of bioactive 5-FU.
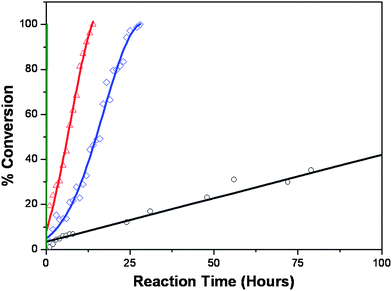 | ||
| Fig. 3 Conversion of ∼1 mM of the 5-FC prodrugs to 5′-DFCR in the presence of 0.5 mg ml−1carboxylesterase: Capecitabine (green), 5-FCOleyl (red △), 5-FCPhytanyl (blue ◇), and 5-FCPalmityl (black ○). Capecitabine, which displays no self-assembly behaviour, is fully converted within 15 minutes, the minimum time required to obtain the first measurement. 5-FCOleyl and 5-FCPhytanyl both produce nanoassemblies with an inverse bicontinuous cubic internal structure. The self-assembly of these prodrugs results in reduced conversion rate, with 5-FCOleyl fully converted after 13 h, and 5-FCPhytanyl after 26 h. The solid–lipid nanoparticle structure of the 5-FCPalmityl prodrug makes it difficult for the enzyme to access the cleavage site, and thus more than a week is needed for full hydrolysis to occur. | ||
The chemotherapeutic efficacy of the 3 different 5-FC drug lipid conjugates was assessed in vivo using a 4T1 breast cancer model in BALB/c mice. Preliminary in vitro studies against several primary cells and tumour cell lines demonstrated that the nanoassemblies possess low toxicity. Initial in vivo studies (n = 6 mice in 13 treatment groups) which examined the three 5-FC lipid prodrugs at a range of concentrations indicated that dosages of 0.5 mmol kg−1 day−1 of 5-FCPhy and 5-FCOle displayed similar efficacy to Capecitabine at the same concentration. A second in vivo study (n = 10 mice in 4 treatment groups) undertaken to maximize the efficacy of 5-FCPhy and 5-FCOle examined these two nanoassemblies at a concentration of 1 mmol kg−1 day−1. After 17 days, the 5-FCOle treatment group possessed significantly smaller tumours than all other treatment groups (Fig. 4A and C). The average tumour size in the 5-FCOle treatment group was ∼1/5 the size of the tumours in both the 5-FCPhy and Capecitabine treatment groups and ∼1/10 the size of tumours in the untreated control group. In addition, spleens in the 5-FCOle treatment group were still normal in size, while spleens from the other treatment groups were enlarged 2–3 times indicating a strong immune response to the induced tumours (Fig. 4D). On day 17, half of the mice in the 5-FCOle group were sacrificed, while treatment was discontinued on the other half. Another full week was required for tumours in this group to reach a comparable average size to the other three treatment groups at day 17. In order to further assess the sustained release profile and efficacy of the 5-FCOle prodrug, a third study (n = 10 mice in 3 treatment groups) was undertaken with mouse xenografts inoculated with the MDA-MB-231 breast tumour. Mice were orally administered the lipid prodrug nanoassemblies at two different dosages (0.75 mmol kg−1 day−1 and 1 mmol kg−1 day−1) via a pre-optimized dosing scheme of two days on/two days off determined in a separate in vivo study. After 17 days of treatment, tumours in both 5-FCOle treatment groups displayed no significant change in tumour volume, while the control group displayed a ∼4 fold increase in volume (Fig. 4B). Since nanoparticle formulations of drugs have been shown to improve drug circulation time, this optimized dosing scheme corresponds well to a prodrug experiencing an increase in prodrug circulation time in conjunction with an increased time scale of initial enzymatic prodrug conversion. The lack of tumour growth seen with this optimized dosing interval combined with the in vitro release profile provides strong evidence of sustained release of the 5-FCOlein vivo.
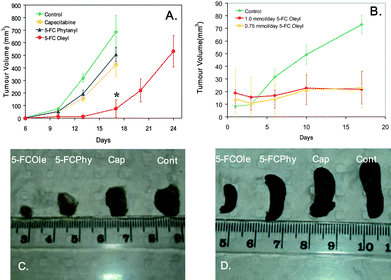 | ||
| Fig. 4 (A) Comparison of 4T1 tumour volume in female BALB/c mice administered 1 mmol per mouse per day of Capecitabine, 5-FCOleyl, 5-FCPhytanyl, and the control group receiving 0.54 mg F127 in water per mouse per day (poloxamer only). After 17 days, tumour volume in the 5-FCOleyl group is significantly smaller than the other treatment groups. *Dosing was discontinued in 5-FCOleyl group on day 17. (B) Comparison of MDA-MB-231 tumour volume in BALB/c-Fox1nu/ARC (athymic nude) administered 0.75 mmol per mouse 5-FCOleyl, 1 mmol per mouse 5-FCOleyl, and the poloxamer only (control) delivered following a dosing regime of 2 days on/2 days off. After 17 days, tumour volume remained unchanged in mice receiving 5-FCOleyl. (C) Comparison of excised 4T1 tumours from BALB/c mice administered 1 mmol per mouse per day of Capecitabine, 5-FCOleyl, 5-FCPhytanyl, and poloxamer only (control). (D) Comparison of excised spleens from BALB/c mice administered 1 mmol per mouse per day of Capecitabine, 5-FCOleyl, 5-FCPhytanyl, and poloxamer only (control). | ||
We have developed highly effective, novel nanomedicines consisting of a series of 5-FU prodrug amphiphiles that can be orally administered. Formulation of these lipid prodrugs into nanostructured nanoparticles enabled the controlled release of the bioactive 5-FU molecule. In addition, we observed marked improvement in efficacy against the mouse 4T1 breast tumour model over a commercially available chemotherapeutic agent (Capecitabine), which has a similar structure but no capacity to self-assemble. This breast cancer model is extremely aggressive, analogous to a stage 4 breast tumour in humans, and the ability of the drug to slow the growth of such tumours is highly significant.25 Furthermore, the 5-FCOle prodrug essentially stopped tumour growth in the human MDA-MB-231 xenograft breast tumour model26,27 demonstrating the significant untapped potential of this form of nanomedicine for progressive cancer therapies.
Methods
Materials
The prodrug Capecitabine and 2′,3′-Di-O-acetyl-5′-deoxy-5-fluorocytidine were purchased from Xingcheng Chemphar Co. Ltd. (P.R.China). Cetyl chloroformate, oleyl alcohol, phytol and all other materials and solvents were purchased from Sigma-Aldrich and used without further modification.Phytanol was synthesised by hydrogenation of phytol according to the reported method without purification of the stereoisomers.28Oleyl alcohol and phytanol were activated by triphosgene to form oleyl chloroformate and phytanyl chloroformate, respectively.29 The chloroformate solution of the respective chains was reacted with 2′,3′-Di-O-acetyl-5′-deoxy-5-fluorocytidinevia a carbamate bond at position N4, followed by deprotection of the acetyl groups on positions 2′and 3′ to yield 5-FCPal, 5-FCOle, and 5-FCPhy, as shown in Fig. 1. Prodrug conjugates were characterized by TLC, HPLC, LC/MS, 1HNMR, 13CNMR and microanalysis.
Carboxylesterase, extracted from porcine liver with 131 units mg−1 (at pH 8.0 and 25 °C), was purchased from Sigma-Aldrich. Milli-Q water was used to prepare the prodrug solutions with appropriate concentrations, while phosphate buffered saline (pH 7.4, PBS) was used in the preparation of the different enzyme solutions.
Preparation of nanoparticle dispersions
Solid–lipid nanoparticle dispersions were prepared by a modification of the methods of Fong et al.30 Briefly, neat 5-FCPal and 10% w/w lipid of F127 were combined and melted at 120 °C. Warm Milli-Q water was added and the mixture was initially dispersed using an Ultra Turrax® homogenizer at 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 min. The coarse dispersion was then sonicated for 3 h followed by high pressure homogenization (EmulsiFlex-C3 Homogenizer, Avestin Inc., Canada) at ∼170 MPa (25000 psi) for 20 min. Dispersions of 5-FCOle and 5-FCPhy were prepared by initially dissolving the neat prodrug and 10% w/w lipid of F127 in a small amount of ethanol. The dissolved mixture was then added drop wise to Milli-Q water. The mixture was then sonicated for two hours followed by extrusion through a series of polycarbonate membranes using a mini-extruder (Avanti Polar Lipids, US). The size distribution (intensity vs. size) of all dispersion was obtained using a Malvern Zetasizer (Nano ZS, Malvern).
000 rpm for 10 min. The coarse dispersion was then sonicated for 3 h followed by high pressure homogenization (EmulsiFlex-C3 Homogenizer, Avestin Inc., Canada) at ∼170 MPa (25000 psi) for 20 min. Dispersions of 5-FCOle and 5-FCPhy were prepared by initially dissolving the neat prodrug and 10% w/w lipid of F127 in a small amount of ethanol. The dissolved mixture was then added drop wise to Milli-Q water. The mixture was then sonicated for two hours followed by extrusion through a series of polycarbonate membranes using a mini-extruder (Avanti Polar Lipids, US). The size distribution (intensity vs. size) of all dispersion was obtained using a Malvern Zetasizer (Nano ZS, Malvern).
Small angle X-ray scattering (SAXS)
Synchrotron SAXS on dispersed nanoparticles was carried out at the SAXS/WAXS beamline of the Australian Synchrotron. The experiments used a beam of wavelength λ = 0.80 Å (15.0 keV) with dimensions 700 µm × 500 µm and a typical flux of 1.2 × 1013photons s−1. 2-D diffraction images were recorded on a Mar CX165 detector. Liquid crystalline mesophases give rise to distinct diffraction patterns which may be used as an unambiguous identification for each phase, provided an adequate number of reflections are observed. Additional synchrotron SAXS was carried out at the 15-ID-D (ChemMatCARS) beamline of the Advanced Photon Source, Argonne, Illinois.31 The experiments used a beam of wavelength λ = 1.50 Å (8.27 keV) with dimensions 200 µm × 100 µm and a typical flux of 1 × 1012photons s−1. 2-D diffraction images were recorded on a Bruker 6000 CCD detector with an active area of 94 × 94 mm2 and a pixel size of 92 µm. The CCD detector was offset from the main beam allowing analysis in the q-range 0.0187–0.807 Å−1 at a sample to detector distance of 0.6 m. Temperature control was in the range 7–90 °C.All images were analysed using AXcess, a custom-built SAXS analysis program written by Dr Andrew Heron: details on the use of AXcess for SAXS analysis are reported in a review article.32
Cryo-transmission electron microscopy (TEM)
Cryo-TEM was carried out using a laboratory-built vitrification system allowing humidity to be kept at close to 90% during sample plunging and vitrification. 4–5 µl of sample solution was placed on a 300 mesh copper TEM grid coated with a lacey carbon film (ProSciTech, Thuringowa Qld 4817Australia), and the resulting thin film was then vitrified by plunging into liquid ethane. Grids were stored in liquid nitrogen before being transferred into a Gatan 626-DH Cryo-holder. Imaging was carried out using either an FEI Tecnai 12 TEM, operating at 120 kV, equipped with a MegaView III CCD camera and Analysis imaging software (Olympus Soft Imaging Solutions) or a Tecnai TF30 TEM fitted with a Gatan US1000 2k × 2k CCD Camera, and Digital Micrograph Imaging software, operating at 200 kV.Molecular modeling
The energy minimum conformation of the molecule was determined by Chem 3D Pro v11.0 (CambridgeSoft Corporation) using its MM2 energy minimization routine.Enzyme hydrolysis
A 1 mg ml−1enzyme solution was then mixed with an equal volume of the prodrug dispersions by gently shaking. The mixture was maintained at 37 °C throughout the course of the experiment, and a sample aliquot was taken as time zero when the enzyme was added to the prodrug solution. The concentration of the hydrolysis product was then measured at set time points using a Thermo Finnigan LC/MS (Finnigan LCQ Series, Thermo Scientific, USA) equipped with an atmospheric pressure chemical ionization (APCI) probe in positive ion mode. Data acquired from Xcalibur Quant chromatography software were used for data acquisition and processing. The decrease in the concentration of the original substrate with time was generated by integration of the peak related to the intact amphiphile at the different time intervals.In vivo experiments
All animal work was performed in accordance with CSIRO animal ethics guidelines under ACEC 07/11 and AEC 09/07. For the 4T1 model, female BALB/c mice were obtained from the Animal Resource Centre (Canning Vale, WA, Australia) at 6–8 weeks of age. After at least 1 week of observation, mice were injected with 5 × 1044T1 cells per 10 µl PBS into the 3rd mammary fat pad on the right hand side. Daily treatment was started 3 days post-inoculation. Approximately 2% of animals did not form tumours throughout the course of the experiment. Mice were divided into 4 groups of 10 and administered the following daily treatments for 21 days calculated based on a 20 g mouse: 1 mmol 5-FCOle, 1 mmol 5-FCPhy, 1 mmol Capecitabine, or a 1.5 mg ml−1 solution of F127 in watervia orogastric gavage. The length and breadth of the tumours were measured and recorded on day 6, 10, 13, and 17 post-tumour inoculations. On day 17 of drug administration, mice in the control, Capecitabine, 5-FCPhy, and half of the 5-FCOle treatment groups were euthanized and blood and organs harvested. Treatment was discontinued in the remaining mice from the 5-FCOle treatment group, and tumour volume was monitored for an additional week with measurements taken on day 20 and 24. Remaining mice were euthanized on day 24.For the MDA-MB-231 model, female BALB/c-Fox1nu/ARC (athymic nude) mice were obtained from the Animal Resource Centre (Canning Vale, WA, Australia) at 4–6 weeks of age. After 1 week of observation, mice were injected with 1 × 105 MDA-MB-231cells per 100 µl equal mixture of PBS and Geltrex (Invitrogen, Vic, Australia) subcutaneously into right flank. Treatment was started ∼3 weeks post-inoculation when tumours were visible in the majority of mice. Approximately 10% of animals did not form tumours throughout the experiment. Mice were divided into 3 groups of 10 and administered the following treatments following a dosing regime of 2 days on/2 days off for 17 days: 1 mmol 5-FCOle, 0.75 mmol 5-Ole, or a 1.5 mg ml−1 solution of F127 in watervia orogastric gavage. The length and breadth of the tumours were measured and recorded on day 1, 3, 6, 10, and 17 of treatment administration. Tumour volume as calculated by the following formula: volume = (breadth × length2)/2. On day 17, all mice were euthanized and organs harvested.
Acknowledgements
We would like to thank Bio21 for the use of the Cryo-TEM equipment and Dr. Kenneth Goldie for his assistance in obtaining the cryo-TEM images. The authors also thank Dr. Greg Warr for discussion pertaining to the work herein. We would like to thank Dr. David Cookson and Dr. Robert Knott for their assistance with experiments carried out at the ChemMatCARS beamline, sector 15 at the Advanced Photon Source. Use of the ChemMatCARS sector 15 at the Advanced Photon Source was supported by the Australian Synchrotron Research Program, which is funded by the Commonwealth of Australia under the Major National Research Facilities Program. ChemMatCARS Sector 15 is principally supported by the National Science Foundation/Department of Energy under grant number CHE-0535644. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357.Part of this research was undertaken on the SAXS/WAXS beamline at the Australian Synchrotron, Victoria, Australia. Calum J. Drummond is the recipient of an Australian Research Council Federation Fellowship.Notes and references
- B. J. Boyd, S. M. Khoo, D. V. Whittaker, G. Davey and C. J. H. Porter, Int. J. Pharm., 2007, 340, 52–60 CrossRef CAS.
- C. J. Drummond and C. Fong, Curr. Opin. Colloid Interface Sci., 1999, 4, 449–456 CrossRef CAS.
- K. Hattori, Y. Kohchi, N. Oikawa, H. Suda, M. Ura, T. Ishikawa, M. Miwa, M. Endoh, H. Eda, H. Tanimura, A. Kawashima, I. Horii, H. Ishitsuka and N. Shimma, Bioorg. Med. Chem. Lett., 2003, 13, 867–872 CrossRef CAS.
- T. Ishikawa, M. Utoh, N. Sawada, M. Nishida, Y. Fukase, F. Sekiguchi and H. Ishitsuka, Biochem. Pharmacol., 1998, 55, 1091–1097 CrossRef CAS.
- F. Kratz, I. A. Muller, C. Ryppa and A. Warnecke, ChemMedChem, 2008, 3, 20–53 CrossRef CAS.
- J. Rautio, H. Kumpulainen, T. Heimbach, R. Oliyai, D. Oh, T. Jarvinen and J. Savolainen, Nat. Rev. Drug Discovery, 2008, 7, 255–270 CrossRef CAS.
- L. H. Reddy, H. Khoury, A. Paci, A. Deroussent, H. Ferreira, C. Dubernet, X. Decleves, M. Besnard, H. Chacun, S. Lepetre-Mouelhi, D. Desmaele, B. Rousseau, C. Laugier, J. C. Cintrat, G. Vassal and P. Couvreur, Drug Metab. Dispos., 2008, 36, 1570–1577 CrossRef CAS.
- L. H. Reddy, J. M. Renoir, V. Marsaud, S. Lepetre-Mouelhi, D. Desmaele and P. Couvreur, Mol. Pharmaceutics, 2009, 6, 1526–1535 CrossRef CAS.
- K. Larsson, J. Dispersion Sci. Technol., 1999, 20, 27–34 CrossRef CAS.
- S. M. Sagnella, C. E. Conn, I. Krodkiewska and C. J. Drummond, Soft Matter, 2009, 5, 4823–4834 RSC.
- S. M. Sagnella, C. E. Conn, I. Krodkiewska, M. Moghaddam, J. M. Seddon and C. J. Drummond, Langmuir, 2010, 26, 3084–3094 CrossRef CAS.
- P. T. Spicer, Curr. Opin. Colloid Interface Sci., 2005, 10, 274–279 CrossRef CAS.
- S. M. Sagnella, C. E. Conn, I. Krodkiewska, M. Moghaddam and C. J. Drummond, J. Phys. Chem. B, 2010, 114, 1729–1737 CrossRef CAS.
- D. L. Gin, C. S. Pecinovsky, J. E. Bara and R. L. Kerr, Liquid Crystalline Functional Assemblies and Their Supramolecular Structures, Springer, 2008, vol. 128, pp. 181–222 Search PubMed.
- J. Lee and I. W. Kellaway, Int. J. Pharm., 2000, 195, 29–33 CrossRef CAS.
- J. Lee and I. W. Kellaway, Drug Dev. Ind. Pharm., 2002, 28, 1155–1162 CrossRef CAS.
- L. Sagalowicz, M. E. Leser, H. J. Watzke and M. Michel, Trends Food Sci. Technol., 2006, 17, 204–214 CrossRef CAS.
- X. Mulet, D. F. Kennedy, C. E. Conn, A. Hawley and C. J. Drummond, Int. J. Pharm., 2010, 395, 290–297 CrossRef CAS.
- H. Ishitsuka, I. Shimma and I. Horii, Yakugaku Zasshi, 1999, 119, 881–897 CAS.
- J. McKendrick and J. Coutsouvelis, Expert Opin. Pharmacother., 2005, 6, 1231–1239 Search PubMed.
- M. Miwa, M. Ura, M. Nishida, N. Sawada, T. Ishikawa, K. Mori, N. Shimma, I. Umeda and H. Ishitsuka, Eur. J. Cancer, 1998, 34, 1274–1281 CrossRef CAS.
- T. Kaasgaard and C. J. Drummond, Phys. Chem. Chem. Phys., 2006, 8, 4957–4975 RSC.
- X. Mulet, X. Gong, L. J. Waddington and C. J. Drummond, ACS Nano, 2009, 3, 2789–2797 CrossRef CAS.
- T. Tabata, M. Katoh, S. Tokudome, M. Nakajima and T. Yokoi, Drug Metab. Dispos., 2004, 32, 1103–1110 CrossRef CAS.
- G. H. Heppner, F. R. Miller and P. V. M. Shekhar, Breast Cancer Res., 2000, 2, 331–334 CrossRef CAS.
- M. Crepin, V. Salle, H. Raux, R. Berger, R. Hamelin, D. Broutyboye and L. Israel, Anticancer Res., 1990, 10, 1661–1666 CAS.
- P. Mullen, A. Ritchie, S. P. Langdon and W. R. Miller, Int. J. Cancer, 1996, 67, 816–820 CrossRef CAS.
- C. J. Burns, L. D. Field, K. Hashimoto, B. J. Petteys, D. D. Ridley and M. Rose, Aust. J. Chem., 1999, 52, 387–394 CrossRef CAS.
- W. Ding and J. S. Fritz, J. Chromatogr., A, 1999, 831, 311–320 CrossRef CAS.
- C. Fong, I. Krodkiewska, D. Wells, B. J. Boyd, J. Booth, S. Bhargava, A. McDowall and P. G. Hartley, Aust. J. Chem., 2005, 58, 683–687 CrossRef CAS.
- D. Cookson, N. Kirby, R. Knott, M. Lee and D. Schultz, J. Synchrotron Radiat., 2006, 13, 440–444 CrossRef CAS.
- J. M. Seddon, A. M. Squires, C. E. Conn, O. Ces, A. J. Heron, X. Mulet, G. C. Shearman and R. H. Templer, Philos. Trans. R. Soc., A, 2006, 364, 2635–2655 CrossRef CAS.
Footnote |
| † S. Sagnella and X. Gong contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2011 |