DOI:
10.1039/C0NR00773K
(Paper)
Nanoscale, 2011,
3, 1149-1157
Preparation of TiO2–Pt hybrid nanofibers and their application for sensitive hydrazine detection†
Received
17th October 2010
, Accepted 17th November 2010
First published on 7th January 2011
Abstract
The TiO2–Pt hybrid nanofibers with an average dia. 72.6 nm were fabricated by electrospinning poly(vinylpyrrolidone)–ethanol solution containing platinum acetate and titanium tetraisopropoxide, followed by calcination in air at 500 °C for 3 h. High resolution TEM showed that Pt nanoparticles with an average diameter of ∼2 nm were well dispersed in the anatase TiO2 nanofibers. Compared to pristine TiO2 nanofibers (average dia. 67.7 nm), the incorporation of Pt nanoparticles into TiO2 nanofibers can greatly enhance the oxidation of hydrazine in neutral solution. The amperometric hydrazine sensor, using the as-prepared TiO2–Pt hybrid nanofibers as the electrochemical catalyst, shows a wide linear range (up to 1.03 mM), a good limit of detection (0.142 µM), and a high sensitivity of 44.42 µA mM−1 cm−2. In addition, the excellent anti-interference property, free of matrix effect from real water samples and good reproducibility make the developed hydrazine sensor promising for real applications.
Introduction
The applications of metal nanoparticles in sensors and biosensors have witnessed a significant growth in the past few years because of the excellent catalytic properties of highly dispersed metal nanoparticles. Among different metal nanoparticles, platinum (Pt) nanoparticles are particularly interesting and have gained considerable attention. Pt nanoparticles have been widely employed in the construction of electrochemical sensors for glucose,1,2L-cysteine,3salicylic acid,4 and hydrogen peroxide.2,5 However, metal nanoparticles including Pt nanoparticles usually suffer from the aggregation issue, which greatly limits their application. To circumvent this problem, several strategies have been developed to stabilize metal nanoparticles, such as immobilization of metal nanoparticles on colloidal particles, carbon nanotubes, and electrospun fibers, or capping by polyelectrolytes or ligands.6,7 Owing to the high porosity and large surface area, nanofibers fabricated viaelectrospinning have been shown as efficient supports for catalytic metal nanoparticles. Electrospinning is a relatively simple and versatile method for fabricating nanofibers, which is originally applied to pure organic polymers.8 Recently, this technique has been extended to produce composite nanofibers by electrospinning polymer solution containing metal salts. Furthermore, these polymer–metal salt composite nanofibers can be employed as the precursor to fabricate one dimensional metal oxide nanofibers by selective removal of polymer matrix and decomposition of metal salt to metal oxide through calcination in air.9 A series of metal oxide nanofibers have been developed following this procedure, such as CuO,10,11ZnO,12Co3O4,9,13TiO2,8,14etc. In order to incorporate Pt nanoparticles into electrospun metal oxide nanofibers, several strategies have been reported. For instant, Kim et al. fabricated Pt-loaded TiO2 nanofibers by electrospinning polyethylene oxide aqueous solutions containing Ti(OH)n slurry and Pt nanoparticles and subsequent calcination of the composite nanofibers at 500 °C for 4 h.15 Formo et al. functionalized TiO2 by a post-treatment of the electrospun TiO2 nanofibers in a polyol reduction bath to coat the surface of anatase nanofibers with Pt nanoparticles.6
In this work, we present a more facile approach to produce TiO2–Pt hybrid nanofibers by a two-step synthetic route. Platinum acetate (Pt(OAc)2) and titanium tetraisopropoxide (Ti(OiPr)4, a sol–gel precursor to titania) were co-dissolved in poly(vinylpyrrolidone) (PVP) solution and electrospun to generate precursor composite nanofibers. After calcination in air at 500 °C for 3 h, polymer matrix was completely degraded and Pt(OAc)2 was decomposed to Pt, thus TiO2–Pt hybrid nanofibers were generated. The as-prepared TiO2–Pt hybrid nanofibers were then applied to modify glassy carbon electrode (GCE) and highly sensitive, selective, and reproducible amperometric detection of hydrazine was achieved.
Experimental
Reagents
PVP (Mw = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300000), Ti(OiPr)4 (97%) and Nafion® perfluorinated resin solution (20 wt% in lower aliphatic alcohols and water) were purchased from Sigma-Aldrich. Pt(OAc)2 (99.9%) was obtained from City Chemical LLC. Ascorbic acid, uric acid, ethanol, hydrazine, and D-(+)-glucose were supplied by Acros Organics. 0.1 M pH 7.0 phosphate buffer solution was prepared using Na2HPO4 and NaH2PO4. All aqueous solutions were prepared using deionized water (18.2 MΩ cm) generated by a Barnstead water system.
300000), Ti(OiPr)4 (97%) and Nafion® perfluorinated resin solution (20 wt% in lower aliphatic alcohols and water) were purchased from Sigma-Aldrich. Pt(OAc)2 (99.9%) was obtained from City Chemical LLC. Ascorbic acid, uric acid, ethanol, hydrazine, and D-(+)-glucose were supplied by Acros Organics. 0.1 M pH 7.0 phosphate buffer solution was prepared using Na2HPO4 and NaH2PO4. All aqueous solutions were prepared using deionized water (18.2 MΩ cm) generated by a Barnstead water system.
Preparation of TiO2–Pt nanofibers
A 0.2 mL of Pt(OAc)2–saturated ethanol was mixed with 0.2 mL of acetic acid and 0.1 mL of Ti(OiPr)4. This solution was then added to 0.5 mL of Pt(OAc)2–saturated ethanol solution containing 0.03 g PVP, followed by 5 min mixing. The final mixture was immediately loaded into a plastic syringe equipped with a 23 gauge needle made of stainless steel. Electrospinning was conducted at an applied voltage of 20 kV with a feeding rate of 0.3 mL h−1 and a collection distance of 5 cm. The precursor nanofibers were then calcined in air at 500 °C for 3 h to generate TiO2–Pt nanofibers. Pure TiO2 nanofibers were also synthesized following a similar procedure.
Preparation of TiO2–Pt nanofiber modified electrode
Prior to the surface modification, glassy carbon electrode (GCE, dia. 3 mm) was polished with 1 μm and 0.05 μm alumina slurries sequentially, and then rinsed with DI water. After that, the electrode was sonicated in ethanol and deionized water, and then dried at room temperature. To prepare 5 mg/mL TiO2–Pt nanofiber suspension, 5 mg TiO2–Pt nanofibers was first suspended in 1 mL ethanol and sonicated for 1 h, and then a 5 μL suspension was dropped onto the surface of GCE. After drying in air, an aliquot of 5 μL Nafion solution (1 wt% in ethanol) was cast on the layer of TiO2–Pt nanofibers in order to entrap the nanofibers. The as-prepared electrode (denoted as TiO2–Pt NFs/GCE) was immersed in water for 1 h to wet the Nafion layer thoroughly before use. Similar procedure was also applied to prepare TiO2 nanofibers modified GCE (TiO2 NFs/GCE) and Nafion modified GCE (Nafion/GCE) which serve as the control electrodes.
Apparatus and electrochemical measurements
A JEOL 6335F field-emission scanning electron microscope (SEM) was employed to examine the morphology and the size of the as-electrospun nanofibers before and after calcination. More detailed morphology and selected area electron diffraction (SAED) patterns were studied using a Tecnai T12 transmission electron microscope (TEM) operated at 120 kV. High resolution TEM images were obtained with a JEOL 2010 FasTEM. XRD patterns and FTIR spectra of the as-prepared samples were obtained with an Oxford diffraction XcaliburTM PX Ultra with ONYX detector and a Nicolet Magna-IR 560 spectrophotometer, respectively. Cyclic voltammetry (CV) measurements were performed on a Model CHI 601C Electrochemical Workstation (CH Instruments, USA). All electrochemical experiments were conducted using a three-electrode electrochemical cell (a working volume of 5 mL) with a working electrode, an Ag/AgCl reference electrode, and a platinum wire counter electrode. For amperometric detection, all measurements were performed by applying an appropriate potential to the working electrode and allowing the transient background current to decay to a steady-state value, before the addition of the analyte. A stirred solution was employed to provide convective transport.
Results and discussions
Characterization of the nanofibers before and after calcination
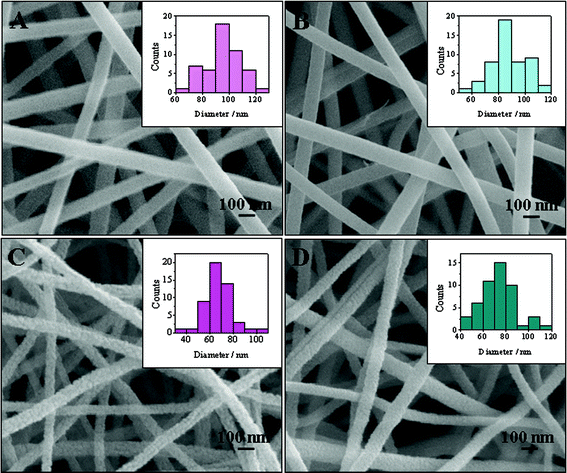
SEM was first employed to investigate the morphology of the precursor nanofibers and the corresponding calcined products. Fig. 1A and B present the typical SEM images of electrospun TiO2–PVP and TiO2–Pt(OAc)2–PVP composite nanofibers, respectively. Since Ti(OiPr)4 can be rapidly hydrolyzed by moisture in the air,8 amorphous TiO2 is present in the electrospun nanofibers. After calcination in air at 500 °C, PVP was completely degraded and the calcined samples remained as the nanofibrous structures (Fig. 1C and D). However, a decrease in the average diameter was observed (Table 1), which could be attributed to the loss of PVP, the crystallization of TiO2, and/or the decomposition of Pt(OAc)2. In addition, the surfaces of calcined nanofibers were not as smooth as the precursor nanofibers. As presented in Fig. 1C and D, TiO2 nanofibers, which were composed of bigger nanoparticles, possessed rougher surface than that of TiO2–Pt nanofibers. The size distribution calculated from 50 randomly selected nanofibers was shown in the inset of Fig. 1.
Table 1 Average diameter of nanofibers before and after calcination
| |
TiO2–PVP |
TiO2 |
TiO2–Pt(OAc)2–PVP |
TiO2–Pt |
| Average diameter/nm |
95.98 ± 13.32 |
67.73 ± 12.12 |
92.10 ± 17.22 |
72.61 ± 15.04 |
 |
| | Fig. 1
SEM images of (A) TiO2–PVP nanofibers, (B) TiO2–Pt(OAc)2–PVP nanofibers, (C) TiO2 nanofibers obtained after calcination, and (D) TiO2–Pt obtained after calcination. Insets: histograms showing the size distribution of nanofibers. Each size distribution was obtained from 50 randomly selected nanofibers. | |
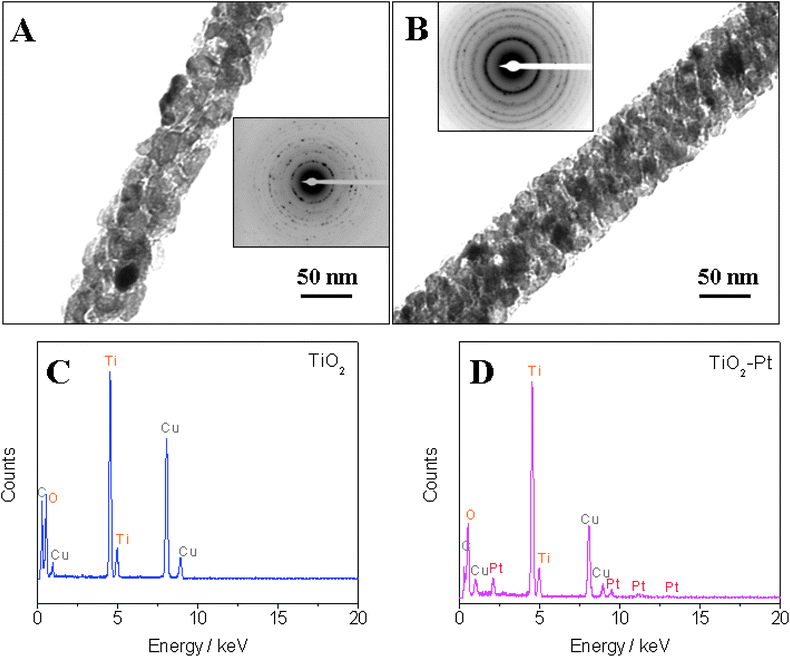
Fig. 2A and B show typical TEM images for single TiO2 nanofiber and TiO2–Pt nanofiber, respectively. One can see that numerous black dots are present on the surface of TiO2 nanofibers or embedded in the TiO2 nanofibers. The condensation of Pt nanoparticles may be the major reason for the formation of these black dots. The insets of Fig. 2A and B display corresponding SAED patterns, indicating the polycrystalline structure of the nanofibers. Furthermore, EDX analysis was conducted with a Tecnai T12 TEM to reveal the difference in compositions of the TiO2 nanofibers and TiO2–Pt hybrid nanofibers. Well-formed peaks for Pt can be observed in the EDX pattern of the TiO2–Pt composite nanofibers (Fig. 2D), while no Pt is found in the TiO2 nanofibers (Fig. 2C).
 |
| | Fig. 2
TEM images and SAED patterns (inset) of (A) TiO2 nanofiber and (B) TiO2–Pt nanofiber. EDX analysis of (C) TiO2 nanofibers and (D) TiO2–Pt nanofibers (C and Cu peaks in EDX come from the copper–carbon grid for TEM). | |
High resolution TEM (HRTEM) was also performed to show the distribution of Pt in the TiO2 nanofibers. As shown in Fig. 3B, small black dots with an average diameter of ∼2 nm are dispersed in the nanofibers. Clear lattice of the black dots cannot be obtained due to the size of the nanofibers and interference from dominant TiO2. A comparison of the EDX analysis carried out at the wide area (Fig. 3C) and the black dot area (Fig. 3D) revealed that the content of Pt in the black dot area is about 4-fold higher than the average content in the whole TiO2–Pt nanofibers (Table 2). This result provides an indirect evidence that the black dots might be condensed Pt nanoparticles. Furthermore, the composition and crystal structure of the TiO2–Pt hybrid nanofibers were characterized by XRD. As demonstrated in Fig. S1†, the dominant anatase phase TiO2 (JCPDS No. 21-1272) is revealed by the diffraction peaks at 2θ values of 25.3, 37.8, 38.6, 48.0, 53.9, 55.1, 62.7, 68.8, and 70.3°, corresponding to (101), (004), (112), (200), (105), (211), (204), (116), and (220) crystal planes, respectively. However, the peaks corresponding to Pt are barely observed in the XRD pattern of TiO2–Pt nanofibers, which may be attributed to the low content of Pt in the hybrid nanofibers. In order to demonstrate that Pt can be generated from the decomposition of Pt(OAc)2, a control experiment was carried out with a suspension containing 6 wt% Pt(OAc)2 and 6 wt% PVP in ethanol. This suspension was electrospun with the same procedure and then the sample was calcined at 500 °C for 3 h. As shown in Fig. S2†, the as-prepared sample exhibits nanofibers structure with an average diameter of 30 nm. As shown in Fig. S3†, XRD pattern of the calcined Pt(OAc)2–PVP nanofibers indicates cubic crystalline structure of Pt (JCPDS No. 04-0802), which is revealed by the diffraction peaks at 2θ values of 39.8, 46.2, 67.5, 81.3, and 85.7°, corresponding to (111), (200), (220), (311), and (222) crystal planes, respectively. This experiment clearly demonstrated the successful conversion of Pt(OAc)2 to Pt by calcination at 500 °C and strongly supported that the black dots observed in HRTEM are Pt nanoparticles.
Table 2 The ratio of different elements in different locations of TiO2–Pt nanofibers
| |
Element |
Atomic% |
| Wide area |
O (K) |
65.3 |
|
Ti (K) |
33.5 |
|
Pt (L) |
1.2 |
| Black dot |
O (K) |
50 |
|
Ti (K) |
43.8 |
|
Pt (L) |
6.2 |
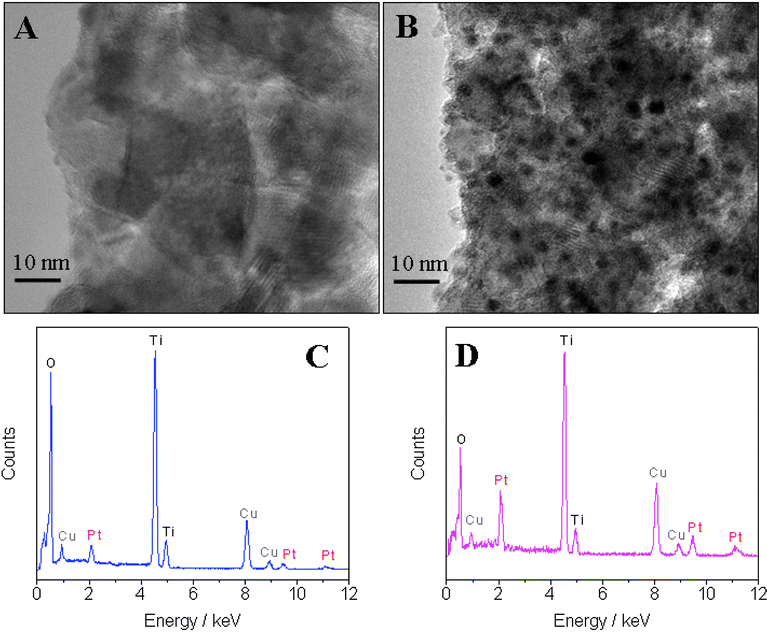 |
| | Fig. 3 Typical HRTEM images of (A) TiO2 nanofibers and (B) TiO2–Pt nanofibers. EDX analysis of (C) the wide area of TiO2–Pt nanofibers and (D) the black dot area in TiO2–Pt nanofibers. C and Cu peaks in EDX come from the copper–carbon grid for TEM. | |
FTIR was further conducted to confirm the complete removal of PVP and conversion of Pt(OAc)2 to Pt. As shown in Fig. 4A, the characteristic peaks of PVP and Pt(OAc)2 (e.g. 3410 cm−1 for O–H, 2960 cm−1 for C–H, 1660 cm−1 for C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, 1660 cm−1 and 1550 cm−1 for symmetric and asymmetric vibrations of acetate, 1430 cm−1 for C–H, 1290 cm−1 for N–OH complex, C–N or C–O, and 847 cm−1 for O–H) significantly weaken or completely disappear, which could be attributed to the degradation of PVP and the decomposition of Pt(OAc)2.13,16–18XPS measurement was also carried out to study the surface compositions of the TiO2–Pt nanofibers. As shown in Fig. 4B, the survey spectrum contains O 1s, Ti 2p and Pt 4d peaks, indicating the surface constitution of the hybrid nanofibers. C 1s peak observed in the survey comes from the background. Based on this typical survey, the weight percentage of Pt is estimated to be around 1.3% (excluding C 1s), in good agreement with the value obtained by EDX analysis (1.2%). A typical high resolution spectrum of Ti 2p region is shown in Fig. 4C, with Ti 2p1/2 and Ti 2p3/2 peaks located at 458.43 and 463.97 eV, respectively. The peak separation of 5.54 eV is in good agreement with the reported literature values.19 On the other hand, the spectrum of Pt 4f region exhibits 4 peaks. The Pt 4f7/2 peak at 71.02 eV and Pt 4f5/2 peak at 74.83 eV with a peak separation of 3.81 eV can be assigned to zero-valent platinum, while the Pt 4f7/2 peak at 73.45 eV and Pt 4f5/2 peak at 76.97 eV with a peak separation of 3.54 eV is ascribed to PtO.20,21 The presence of PtO on the surface of Pt nanostructures is not unusual, since oxygen chemisorption can easily occur at step and kink sites present on the surface of Pt.22
O, 1660 cm−1 and 1550 cm−1 for symmetric and asymmetric vibrations of acetate, 1430 cm−1 for C–H, 1290 cm−1 for N–OH complex, C–N or C–O, and 847 cm−1 for O–H) significantly weaken or completely disappear, which could be attributed to the degradation of PVP and the decomposition of Pt(OAc)2.13,16–18XPS measurement was also carried out to study the surface compositions of the TiO2–Pt nanofibers. As shown in Fig. 4B, the survey spectrum contains O 1s, Ti 2p and Pt 4d peaks, indicating the surface constitution of the hybrid nanofibers. C 1s peak observed in the survey comes from the background. Based on this typical survey, the weight percentage of Pt is estimated to be around 1.3% (excluding C 1s), in good agreement with the value obtained by EDX analysis (1.2%). A typical high resolution spectrum of Ti 2p region is shown in Fig. 4C, with Ti 2p1/2 and Ti 2p3/2 peaks located at 458.43 and 463.97 eV, respectively. The peak separation of 5.54 eV is in good agreement with the reported literature values.19 On the other hand, the spectrum of Pt 4f region exhibits 4 peaks. The Pt 4f7/2 peak at 71.02 eV and Pt 4f5/2 peak at 74.83 eV with a peak separation of 3.81 eV can be assigned to zero-valent platinum, while the Pt 4f7/2 peak at 73.45 eV and Pt 4f5/2 peak at 76.97 eV with a peak separation of 3.54 eV is ascribed to PtO.20,21 The presence of PtO on the surface of Pt nanostructures is not unusual, since oxygen chemisorption can easily occur at step and kink sites present on the surface of Pt.22
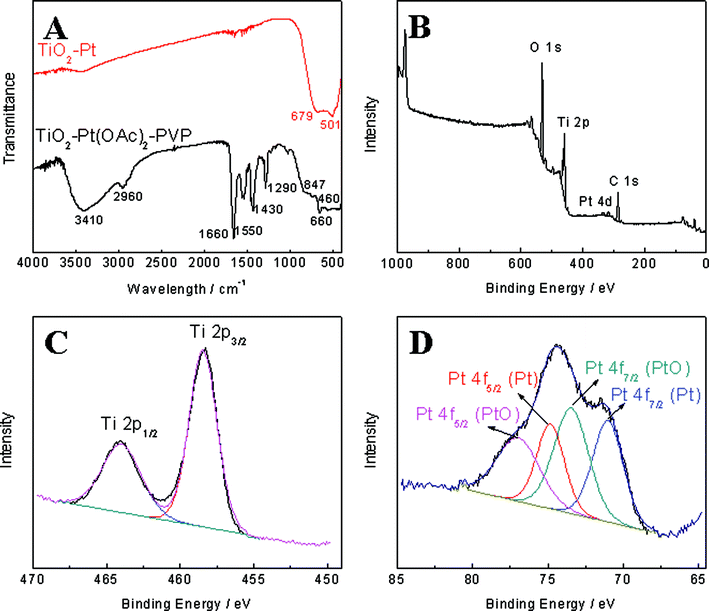 |
| | Fig. 4 (A) FTIR spectra of TiO2–Pt(OAc)2–PVP nanofibers and TiO2–Pt nanofibers. (B) XPS survey spectrum of TiO2–Pt nanofibers. (C) High resolution XPS spectrum of Ti 2p region for the TiO2–Pt nanofibers. (D) High resolution XPS spectrum of Pt 4f region for the TiO2–Pt nanofibers. | |
Electrochemical behavior of TiO2–Pt nanofiber and TiO2 nanofiber modified GCEs on hydrazine oxidation
Hydrazine is widely used in many fields such as rocket fuels, missile systems, weapons of mass destruction, fuel cells, corrosive inhibitors, pesticides, blowing agents, pharmaceutical intermediates, photographic chemical, etc.23–26 However, it is a neurotoxin and has carcinogenic and mutagenic effects.27 Therefore, sensitive and reliable detection of hydrazine is of practical importance to public safety and homeland security.
In order to evaluate the electrocatalytic property of the as-prepared TiO2–Pt NFs towards the oxidation of hydrazine, the CVs in the absence and presence of 2 mM and 4 mM hydrazine were recorded at Nafion/GCE, TiO2 NFs/GCE and TiO2–Pt NFs/GCE and shown in Fig. 5. No oxidation peak was observed at either TiO2 NFs/GCE or Nafion/GCE. However, the oxidation response of hydrazine at TiO2 NFs/GCE was slightly stronger than that at Nafion/GCE even though there is no significant improvement on the onset potential for hydrazine oxidation. Thus TiO2 itself may not be a good catalyst for the hydrazine electrooxidation, but the nanofibrous structure could provide high specific surface area and allow the access of the analytes with minimal diffusion resistance. In addition, TiO2 has an isoelectric point (IEP) in the range from 4 to 6.28 In 0.1 M pH 7.0 phosphate buffer, the surface of TiO2 nanofibers would be negatively charged, which may favor the attachment of protonated hydrazine (N2H5+) onto the nanofibers. Thus the improvement of the electrocatalytic behavior of TiO2 NFs/GCE compared to Nafion/GCE may be attributed to either the enlarged electrode surface area or the favorable adsorption of hydrazine, or their combination effects. In the case of the TiO2–Pt NFs/GCE, a well-defined oxidation peak of hydrazine was exhibited at around 0.55 V, accompanied by a much lower oxidation onset potential and higher oxidation current. The lower onset potential indicated that the catalytic activity of TiO2–Pt NFs is much stronger than that of TiO2 NFs or GCE. The increase of the current response can be attributed to the increase of the electron transfer process and the enhanced electrocatalytic activity. These improvements are obviously ascribed to Pt nanoparticles in the TiO2 nanofibers because of their superior catalytic activity towards the oxidation of hydrazine.
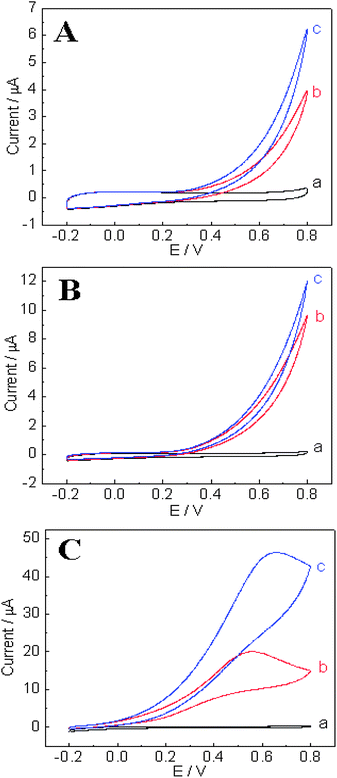 |
| | Fig. 5
CVs recorded at (A) Nafion/GCE, (B) TiO2 NFs/GCE and (C) TiO2–Pt NFs/GCE in 0.1 M pH 7.0 phosphate buffer in the absence (a) and presence of 2 mM (b) and 4 mM (c) hydrazine, respectively. Scan rate is 100 mV s−1. | |
Amperometric detection of hydrazine at the modified electrode
In order to determine the optimum applied potential for the amperometric detection of hydrazine, hydrodynamic voltammogram behavior of the TiO2–Pt NFs/GCE was then investigated by measuring its amperometric responses to 80 µM hydrazine at different applied potentials ranging from 0.2 to 0.8 V. The response for each injection was determined when the current reached 95% of steady-state current. As presented in Fig. 6A and B, the oxidation current response of hydrazine at the TiO2–Pt NFs/GCE initially increases with the increase of applied potential and then gradually reaches a plateau after 0.6 V. This observation is in good agreement with the CV results presented in Fig. 5C, which shows the oxidation peak potential located at 0.55 V. However, the background noises always increase with increased applied potentials. As the detection limit of a sensor depends on both the signal and the background noise level, the signal-to-background noise (S/N) ratios at different applied potentials were calculated and used to determine the optimum applied potential for amperometric detection of hydrazine. Fig. 6B displays the signal as well as S/N ratio against the applied potential. One can see that, at an applied potential of 0.2 V, the highest S/N value could be obtained, but with a very low signal. When the applied potential is higher than 0.5 V, the signal to hydrazine oxidation is impressive; however, the background noise level is very high, thus small S/N ratio was observed which was not favorable for amperometric detection of hydrazine. By trading off the signal and the noise, an applied potential of 0.5 V was selected as the optimum applied potential for subsequent amperometric detection of hydrazine.
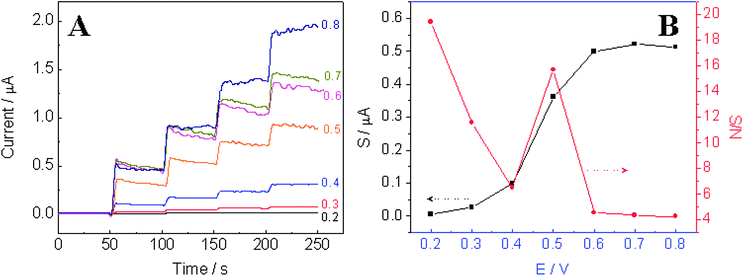 |
| | Fig. 6 (A) Amperometric response of the TiO2–Pt NFs/GCE with four successive additions of 80 µM hydrazine to 0.1 M pH 7 phosphate buffer at different applied potentials ranging from 0.2 V to 0.8 V. (B) Plots of signal and signal-to-background noise ratio against the applied potential. | |
Fig. 7A exhibits the typical amperometric response of the TiO2–Pt nanofibers to successive additions of hydrazine at an applied potential of 0.5 V. Due to the strong oxidation of hydrazine on the TiO2–Pt nanofibers (N2H5+→ N2+ 5H++ 4e-), a rapid increase in the current was noticed after each addition of hydrazine into the supporting electrolyte solution, and a steady-state response was obtained within 3 s, demonstrating fast, stable and efficient catalytic ability of the TiO2–Pt NFs. As a comparison, current response obtained at the TiO2 NFs/GCE was much smaller than that obtained at the TiO2–Pt NFs/GCE, which indicates the superb catalytic ability of Pt nanoparticles. The ultrathin TiO2 nanofibers not only provided a suitable support for the dispersion of Pt nanoparticles and enlarged surface area, but also favored the adsorption of N2H5+, thus bringing N2H5+ closer to the catalytic sites of Pt nanoparticles and then enhancing the efficiency of hydrazine oxidation. Fig. 7B shows the corresponding calibration curves at the TiO2–Pt NFs/GCE and TiO2 NFs/GCE, respectively. The analytical characteristics of both TiO2 NFs/GCE and TiO2–Pt NFs/GCE such as the linear range, sensitivity, and the limit of detection (S/N = 3) were compared and presented in Table 3. The TiO2–Pt NFs/GCE exhibits a sensitivity of 44.42 µA mM−1 cm−2, which is almost 9.6-fold higher than that of TiO2 NFs/GCE. The improvements in the linear range and the limit of detection are also significant. Compared to the values reported in literature, the detection limit of 0.142 µM obtained at the TiO2–Pt NFs/GCE in neutral pH is not as good as that of 200 pM at Au nanoparticles self-assembled on silicate network operated in pH 9.2 (because OH− strongly favors hydrazine oxidation),29 but is comparable to 0.1 µM at carbon on ZnO nanorod array electrode30 and lower than that of 0.5 µM at nano-Au/porous-TiO2 composites,31 1.8 µM at Pd plated boron-doped diamond microdisc array,32 2.9 µM at Pd nanoparticles/carbon nanofibers,33 3.6 µM at overoxidized polypyrrole,34 10 µM at nickel tetrasulfonated phthalocyanine,35etc.
Table 3 Comparison of TiO2–Pt NFs/GCE with TiO2 NFs/GCE
| |
Linear range/mM |
Sensitivity/µA mM−1 cm−2 |
Detection limit/µM |
|
TiO2–Pt NFs/GCE |
1.03 |
44.42 |
0.142 |
|
TiO2
NFs/GCE |
0.16 |
4.62 |
1.52 |
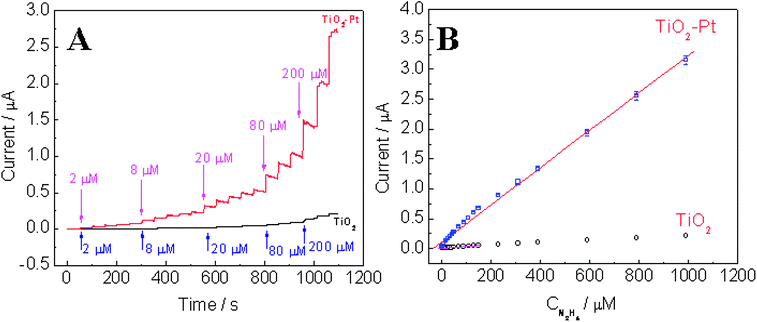 |
| | Fig. 7 (A) Amperometric response of the TiO2–Pt NFs/GCE and TiO2 NFs/GCE with successive additions of hydrazine to 0.1 M pH 7 phosphate buffer at an applied potential of +0.5 V. (B) The corresponding calibration curves with fitting curve and linear range. | |
Anti-interference property and reproducibility study
The selectivity of the hydrazine sensor was evaluated against a range of compounds such as ascorbic acid (AA), uric acid (UA), and glucose (Glu) by holding the TiO2–Pt NFs/GCE at an applied potential of 0.5 V and injecting 8 µM of N2H4, Glu, AA, and UA, sequentially. In addition, water samples obtained from the Mirror Lake and Swan Lake at the University of Connecticut were tested to access the matrix effect due to naturally occurring compounds in real-world samples. As presented in Fig. 8, the as-developed hydrazine sensor exhibits negligible response to AA, UA, and Glu. It has been reported that UA and AA are highly electroactive and can be easily oxidized at the applied potential of 0.5 V. The excellent selectivity against UA and AA may be attributed to the “repelling effect” between negatively charged TiO2 nanofibers and negatively charged AA and UA (deprotonated) in neutral pH, thus AA and UA have minimal chance to be oxidized on the electrode surface. Furthermore, no interference was observed from the injection of lake water samples, indicating that there is no matrix effect from naturally occurred compounds in real water samples. After successive additions of different analytes, the further addition of 8 µM hydrazine shows almost the same response as that observed for the first addition of 8 µM hydrazine, indicating that neither the presence of interference compounds (UA, AA, and Glu) nor naturally occurred species in lake water samples would affect the performance of the TiO2–Pt NFs/GCE. The good selectivity demonstrates the potential utility and feasibility of the developed sensor for hydrazine contaminated environmental samples.
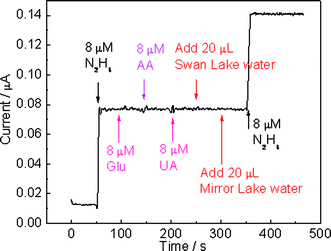 |
| | Fig. 8 Amperometric response of TiO2–Pt NFs/GCE with successive additions of analytes in the sequence of 8 µM hydrazine, 8 µM Glu, 8 µM AA, 8 µM UA, 20 µL water from Swan Lake, 20 µL water from Mirror Lake, and 8 µM hydrazine to 0.1 M pH 7 phosphate buffer at an applied potential of +0.5 V (8 µM is the final concentration of analyte in the electrolyte after considering the dilution effect). | |
Reproducibility of the TiO2–Pt nanofiber modified electrodes was also investigated at 0.5 V. The low relative standard deviation of 3.38% (n = 8) for 80 µM hydrazine demonstrates the high precision of analysis. Additionally, a low relative standard deviation of 7.38% (n = 6) in the response of six TiO2–Pt NFs/GCEs prepared using the same procedure to 80 µM hydrazine shows an excellent electrode-to-electrode reproducibility.
Conclusions
Following a facile two-step synthetic route, TiO2–Pt hybrid nanofibers were prepared by electrospinning and subsequent calcination. SEM and TEM were employed to investigate the morphologies of nanofibers before and after calcination. FTIR was used to confirm the degradation of PVP and the decomposition of Pt(OAc)2. XRD patterns indicated the anatase crystalline property of TiO2 and the conversion of Pt(OAc)2 to Pt. XPS was performed to study the surface composition of the calcined products. The as-prepared TiO2–Pt nanofibers were applied in the hydrazine detection and their electrocatalytic activity was compared with pristine TiO2 nanofibers. Significant enhancement in the hydrazine electrooxidation was achieved with the incorporation of Pt nanoparticles into TiO2 nanofibers, demonstrated by a wider linear range, a lower limit of detection and a higher sensitivity. In addition, the TiO2–Pt hybrid nanofibers showed an excellent anti-interference property towards the electroactive compounds (e.g. AA, UA) and glucose, and good intra-electrode and inter-electrode reproducibility. The matrix effect study using lake water samples further revealed potential practical applications of this sensor for hydrazine detection.
Acknowledgements
We greatly appreciate the funding from NSF and DOE. YD also acknowledges the supports from the UConn CESE through the Graduate Student Research Assistantship Program in Support of Multidisciplinary Environmental Activities by Faculty Members.
References
- L. Q. Rong, C. Yang, Q. Y. Qian and X. H. Xia, Talanta, 2007, 72, 819–824 CrossRef CAS.
- X. J. Bo, J. C. Ndamanisha, J. Bai and L. P. Guo, Talanta, 2010, 82, 85–91 CrossRef CAS.
- L.-P. Liu, Z.-J. Yin and Z.-S. Yang, Bioelectrochemistry, 2010, 79, 84–89 CrossRef CAS.
- Z. Wang, F. Ai, Q. Xu, Q. Yang, J.-H. Yu, W.-H. Huang and Y.-D. Zhao, Colloids Surf., B, 2009, 76, 370–374.
- X. L. Cui, Z. Z. Li, Y. C. Yang, W. Zhang and Q. F. Wang, Electroanalysis, 2008, 20, 970–975 CrossRef CAS.
- E. Formo, E. Lee, D. Campbell and Y. N. Xia, Nano Lett., 2008, 8, 668–672 CrossRef CAS.
- M. H. Yang, Y. H. Yang, Y. L. Liu, G. L. Shen and R. Q. Yu, Biosens. Bioelectron., 2006, 21, 1125–1131 CrossRef CAS.
- D. Li and Y. N. Xia, Nano Lett., 2003, 3, 555–560 CrossRef CAS.
- Y. Ding, Y. Wang, L. Su, M. Bellagamba, H. Zhang and Y. Lei, Biosens. Bioelectron., 2010, 26, 542–548 CrossRef CAS.
- H. Wu, D. D. Lin and W. Pan, Appl. Phys. Lett., 2006, 89, 133125 CrossRef.
- W. Wang, L. Zhang, S. Tong, X. Li and W. Song, Biosens. Bioelectron., 2009, 25, 708–714 CrossRef CAS.
- X. H. Yang, C. L. Shao, H. Y. Guan, X. L. Li and H. Gong, Inorg. Chem. Commun., 2004, 7, 176–178 CrossRef CAS.
- H. Y. Guan, C. L. Shao, S. B. Wen, B. Chen, J. Gong and X. H. Yang, Mater. Chem. Phys., 2003, 82, 1002–1006 CrossRef CAS.
- W. Nuansing, S. Ninmuang, W. Jarernboon, S. Maensiri and S. Seraphin, Mater. Sci. Eng., B, 2006, 131, 147–155 CrossRef CAS.
- H. Kim, Y. Choi, N. Kanuka, H. Kinoshita, T. Nishiyama and T. Usami, Appl. Catal., A, 2009, 352, 265–270 CrossRef CAS.
- Q. Z. Cui, X. T. Dong, J. X. Wang and M. Li, J. Rare Earths, 2008, 26, 664–669 CrossRef.
- Y. Liu, W. Ren, L. Y. Zhang and X. Yao, Thin Solid Films, 1999, 353, 124–128 CrossRef CAS.
- B. Ding, H. Kim, C. Kim, M. Khil and S. Park, Nanotechnology, 2003, 14, 532–537 CrossRef CAS.
- B. Erdem, R. A. Hunsicker, G. W. Simmons, E. D. Sudol, V. L. Dimonie and M. S. El-Aasser, Langmuir, 2001, 17, 2664–2669 CrossRef CAS.
- A. K. Shukla, M. Neergat, P. Bera, V. Jayaram and M. S. Hegde, J. Electroanal. Chem., 2001, 504, 111–119 CrossRef CAS.
- A. K. Shukla, A. S. Arico, K. M. El-Khatib, H. Kim, P. L. Antonucci and V. Antonucci, Appl. Surf. Sci., 1999, 137, 20–29 CrossRef CAS.
- A. S. Arico, A. K. Shukla, H. Kim, S. Park, M. Min and V. Antonucci, Appl. Surf. Sci., 2001, 172, 33–40 CrossRef CAS.
- R. Baron, B. Sljukic, C. Salter, A. Crossley and R. G. Compton, Electroanalysis, 2007, 19, 1062–1068 CrossRef CAS.
- A. Umar, M. M. Rahman and Y. B. Hahn, Talanta, 2009, 77, 1376–1380 CrossRef CAS.
- G. F. Wang, A. X. Gu, W. Wang, Y. Wei, J. J. Wu, G. Z. Wang, X. J. Zhang and B. Fang, Electrochem. Commun., 2009, 11, 631–634 CrossRef CAS.
- J. Wang and L. Chen, Anal. Chem., 1995, 67, 3824–3827 CrossRef CAS.
- J. Liu, W. H. Zhou, T. Y. You, F. L. Li, E. K. Wang and S. J. Dong, Anal. Chem., 1996, 68, 3350–3353 CrossRef CAS.
- J. A. Lewis, J. Am. Ceram. Soc., 2000, 83, 2341–2359 CAS.
- B. K. Jena and C. R. Raj, J. Phys. Chem. C, 2007, 111, 6228–6232 CrossRef CAS.
- J. P. Liu, Y. Y. Li, J. A. Jiang and X. T. Huang, Dalton Trans., 2010, 39, 8693–8697 RSC.
- G. Wang, C. Zhang, X. He, Z. Li, X. Zhang, L. Wang and B. Fang, Electrochim. Acta, 2010, 55, 7204–7210 CrossRef CAS.
- C. Batchelor-McAuley, C. E. Banks, A. O. Simm, T. G. J. Jones and R. G. Compton, Analyst, 2006, 131, 106–110 RSC.
- H. J. Zhang, J. S. Huang, H. Q. Hou and T. Y. You, Electroanalysis, 2009, 21, 1869–1874 CrossRef.
- M. R. Majidi, A. Jouyban and K. Asadpour-Zeynali, Electrochim. Acta, 2007, 52, 6248–6253 CrossRef CAS.
- E. F. Perez, G. D. Neto, A. A. Tanaka and L. T. Kubota, Electroanalysis, 1998, 10, 111–115 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: XRD patterns for TiO2–Pt nanofibers and Pt nanofibers, and SEM images of Pt nanofibers. See DOI: 10.1039/c0nr00773k |
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.