Carbonate-coordinated metal complexes precede the formation of liquid amorphous mineral emulsions of divalent metal carbonates†
Stephan E.
Wolf
a,
Lars
Müller
a,
Raul
Barrea
b,
Christopher J.
Kampf
a,
Jork
Leiterer
c,
Ulrich
Panne
cd,
Thorsten
Hoffmann
a,
Franziska
Emmerling
*c and
Wolfgang
Tremel
*a
aInstitut für Anorganische Chemie und Analytische Chemie, Johannes Gutenberg-Universität, Duesbergweg 10-14, D-55099, Mainz, Germany. E-mail: tremel@uni-mainz.de; Fax: +49 6131 39-25605; Tel: +49 6131 39-25135stephan.eckhard.wolf@gmail.com
bArgonne National Laboratory, Advanced Photon Source, BioCAT, Argonne, Illinois 60439, USA
cBAM Federal Institute of Materials Research and Testing, Richard-Willstätter-Straße 11, D-12489, Berlin, Germany. E-mail: franziska.emmerling@bam.de
dHumboldt-Universität zu Berlin, Institut für Chemie, Brook-Taylor Straße 2, Berlin
First published on 10th January 2011
Abstract
During the mineralisation of metal carbonates MCO3 (M = Ca, Sr, Ba, Mn, Cd, Pb) liquid-like amorphous intermediates emerge. These intermediates that form via a liquid/liquid phase separation behave like a classical emulsion and are stabilized electrostatically. The occurrence of these intermediates is attributed to the formation of highly hydrated networks whose stability is mainly based on weak interactions and the variability of the metal-containing pre-critical clusters. Their existence and compositional freedom are evidenced by electrospray ionization mass spectrometry (ESI-MS). Liquid intermediates in non-classical crystallisation pathways seem to be more common than assumed.
Introduction
Calcium carbonate is a common mineral whose scientific relevance is as widespread as its abundance: it takes a pivotal role in geosciences, biology and industrial applications. Coral reefs and other geological chalk deposits bind an immense amount of carbon dioxide and thus regulate strongly our climate.1 For industrial applications, the effective inhibition of scale incrustation is still a demanding question because scale may cause severe problems, such as impedance of heat transfer, increase of energy consumption, and unscheduled equipment shutdown.2 Calcium carbonate serves further as a filler in plastics and for paper production, as an extender in paints or in drilling fluids for the oil industry.3–5The formation of calcium carbonate has been studied for more than a century and especially its biogenic formation was intensively investigated during the past two decades. Still little is known about the early stages of calcium carbonate crystallization and the associated biomineral formation. In biomineralization we perpetually encounter crystal morphologies and crystallization processes that challenge the classical concepts of crystallization.6 In the past decade, a number of crystallisation paths emerged that do not fit the classical terms of nucleation and crystal growth.7 In the classical model, nucleation is a stochastic clustering process, in which the newly formed crystalline particles are unstable and re-dissolve until they reach a critical particle size.8–10 Nanoparticle-based self-assembly processes (e.g. mesocrystal formation11 or oriented-attachment12,13) and the ubiquitous appearance of amorphous calcium carbonate in (bio)mineralization disprove the simplicity of the classical picture.
Cölfen and Antonietti, who developed and applied the concept of mesocrystallinity in great detail,7 could demonstrate recently that stable calcium carbonate pre-nucleation clusters (PNCs) exist which form in undersaturated solutions of calcium carbonate at pH = 9–10.14 These preformed clusters are proposed to take a relevant role in the formation of calcium carbonate and other biominerals because they may imprint their molecular structure on the growing (bio)mineral.15,16 No structural characterization of the very initial state is currently available; the applied analytical methods do not allow explicit statements on the molecular structure of the pre-nucleation clusters.
Amorphous calcium carbonate (ACC) is assumed to be an intermediate in many biomineralization and biomimetic crystallisation processes.17 It is a transient post-nucleation species with variable short-range order whose structure resembles that of the evolving crystalline calcium carbonate phase.16,18 Up to now, amorphous carbonates of strontium and barium have only been obtained in the presence of additives,19–21 and amorphous phases of the divalent metal carbonates (except for zinc carbonate) have not been described.22 Liquid precursors are an exceptional subtype of these amorphous intermediates which form directly after contact of the educts and maybe via spinodal decomposition. Although liquid precursors are quite common for proteins and polymers,23–27 their existence for inorganic minerals could be demonstrated unequivocally only for calcium carbonate so far.28
Gower et al. were the first to report the induction of a liquid-like intermediate of calcium carbonate with the aid of small amounts of acidic polyelectrolytes and dubbed this intermediate ‘polymer induced liquid precursor’ (PILP).30–32 They could recently expand their concept of PILPs to other minerals such as calcium phosphate, barium carbonate and strontium carbonate.20 The findings of Gower et al. correspond to the claim of Faatz et al.29 that calcium carbonate forms via a hypothetical binodal liquid/liquid phase separation. Rieger et al.33 reported the formation of emulsion-like particles during rapid mixing at high supersaturation at pH = 11. However, it remained unclear whether an instantaneous reaction at the interfaces of the two reactants was achieved. An interfacial reaction of the turbulent liquid phase before complete mixing would lead to an artefact which falsely hints at an emulsion-like precursor.34
In our recent study, the liquid/liquid phase separation of calcium carbonate was shown to occur even in the absence of any additives.28 In order to achieve a large homogeneous supersaturation of the solution and to suppress nucleation by the action of foreign bodies (e.g. macromolecules, spectator ions, liquid/liquid- or solid/liquid-interfaces like vessel walls or due to mixing processes), the crystallisation experiments were carried out in an ultrasonic levitator. Acoustic levitation allows the handling of small samples in a contact-free manner. The sample is held in its position by the axial radiation pressure and radial Bernoulli stress of a standing ultrasonic wave.35,36 The solution/air interface is the only remaining phase boundary; no other contact or foreign materials are present. The concentration of the solute increases in three orders of magnitude by solvent evaporation. Starting from saturated calcium bicarbonate solutions, a liquid CaCO3 precursor was observed to form after several minutes. This liquid amorphous intermediate solidified in the course of time and yielded ACC. Finally, crystalline calcite was formed by heterogeneous nucleation on the solid ACC particles. By cryogenic scanning electron microscopy (cryo-SEM), it was affirmed that the observed liquid particles formed homogenously within the droplet and not by heterogeneous nucleation at the air/water interface. The occurrence of this unusual mineral phase was attributed to the presence of numerous species of carbonate-coordinated calcium-centred complexes which exist already in the undersaturated educt solution. The precipitation was conducted at neutral pH, i.e. at pH ≈ pKA1 = 6.3 where high fractions of protonated carbonate species are present.37 As a result, a variety of carbonate-, bicarbonate- and aquo-coordinated calcium complexes can assumed to be involved in the crystallization equilibria. The presence of several potential bonding partners, variable coordination geometries and coordination numbers of the carbonate groups and the associated distribution of local structures thus favour the formation of a non-crystalline phase.
In this contribution, we investigate the chemical and physicochemical stabilization and formation mechanisms of such mineral emulsions. Further, we provide mass spectrometric evidence for the existence of pre-critical carbonato-coordinated complexes.
Results
Liquid precursors of six different metal carbonates
In order to probe the hypothesis that the formation of a liquid-like CaCO3 mineral phase is mainly due to the complex protonation equilibrium of the carbonate anion at pKA1,‡§ we conducted analogous experiments with carbonates of alkaline earth and other bivalent metals: strontium carbonate (SrCO3), barium carbonate (BaCO3), manganese carbonate (MnCO3), lead carbonate (PbCO3) and cadmium carbonate (CdCO3). Liquid-like particles could be obtained for all samples as shown in Fig. 1. The low contrast variation inside of the particles indicates their liquid-like state; solid spherical particles would show a distinct increase in contrast from the surface to the center of the particles (see Fig. S2, ESI†). An experimental artifact lent further convincing support for their liquid state: during the preparation of the TEM specimens, excess mother solution had to be removed. The solvent removal occasionally exerted a flow on the settled fluid mineral particles and led to coalescence (see Fig. 2a).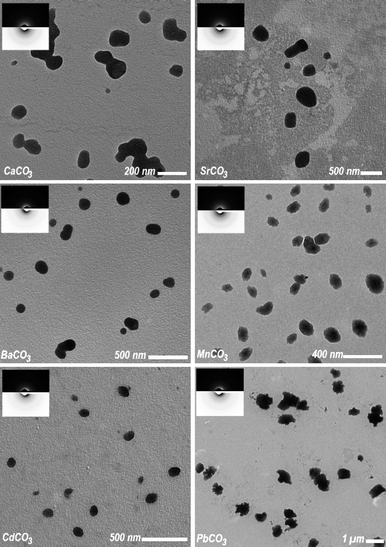 | ||
| Fig. 1 Transmission electron micrographs of liquid-like particles formed by metal carbonates MCO3 (M = Ca, Sr, Ba, Mn, Cd, Pb). The samples were taken at different times: CaCO3 at 400 s; SrCO3 at 120 s; BaCO3 at 400 s; MnCO3 at 600 s; CdCO3 at 800 s; and PbCO3 at 200 s. The respective electron diffraction is shown in the inset which indicates the amorphous state of the particles. The particles seem to occasionally aggregate because of sheer strain which occurs during sample preparation for TEM; a very severe case is depicted in Fig. 2a. Micrographs at higher resolution are provided in Fig. S1 in the ESI†. | ||
 | ||
| Fig. 2 (a) Liquid-like particles of the liquid-like BaCO3 mineral phase which underwent coalescence due to an external forced flow during sample preparation. (b) A liquid CaCO3 intermediate prepared in the presence of 15 mM L−1 NaCl suffered from aggregation and coalescence, e.g. the magnification inset shows an aggregate of four droplets. This behavior points to an electrostatic stabilization of the emulsion. | ||
The lifespan of these mineral emulsions was assessed roughly by TEM by taking samples after different periods of levitation and estimating the time until the first crystalline particles occurred (see Table 1). Cadmium carbonate has the longest lifespan of at least 800 s, whereas strontium carbonate leaves its emulsion-like state relatively fast (250 s). For all six compounds, crystallization of the corresponding metal carbonate could be induced by exposing the liquid-like particles to a highly focused electron beam. The beam-induced crystallisation is attributed to the loss of water of hydration by co-action of radiation heat and ultrahigh vacuum.
| Alkaline earth carbonates | Lifespan | Non-alkaline earth carbonates | Lifespan |
|---|---|---|---|
| CaCO3 | 400 s | MnCO3 | 600 s |
| BaCO3 | 400 s | PbCO3 | 400 s |
| SrCO3 | 250 s | CdCO3 | >800 s |
The formation of the crystalline carbonate phases from the liquid intermediate was monitored in situ by wide-angle scattering (WAXS). The first patterns showed only diffuse scattering of water, which vanished gradually as the water evaporated. The Bragg reflections of the mineral appeared according to their intensity, e.g. the (104) reflection of calcite for CaCO3 or the (111) reflection of strontianite (SrCO3) and witherite (BaCO3, see Fig. S3†) as the first reflections. Cadmium carbonate did not transform to a crystalline mineral phase in detectable amounts during the levitation process, even after the solvent had evaporated completely (cf. Fig. S3†). This is consistent with the long lifespan of the liquid amorphous intermediate of CdCO3 estimated by TEM.
In all cases, the liquid-like particles do not severely aggregate or coalesce although this should be favored energetically because it would reduce the interfacial energy. This implies that the mineral emulsions are weakly stabilized electrostatically, as no other colloidal stabilisation mechanisms such as depletion or steric stabilisation can apply: no additives were employed in these experiments. In classical emulsions like oil/water emulsions, the presence of spectator ions reduces the electrostatic repulsion forces and screens the surface potential in such a way that aggregation or coalescence can occur.38 In our experiments, which start from a pure and saturated calcium bicarbonate solution, no spectator ions are present, whereas in almost all previous studies of the very early formation states of CaCO3 such counter ions were present in high concentrations as a consequence of the preparation procedure (e.g. Na+ or Cl− from mixing of Na2CO3 with CaCl2 solutions). No other additives, such as polymers, were used in the experiments. Therefore, it seems evident that the mineral emulsions can only be stabilized electrostatically. These conclusions are substantiated by experiments in which the liquid calcium carbonate intermediate was prepared in the presence of additional 15 mM L−1 of NaCl. In the presence of NaCl, the CaCO3 emulsion suffers from aggregation and coalescence as expected (see Fig. 2b).
Equilibrium calcium complex species traced by ESI-MS
As outlined above, we assume the formation of different calcium complexes in which water and carbonate species act as a ligand. Electrospray ionization mass spectrometry (ESI-MS)39,40 evolved in the past twenty years as a new method which allows an estimation of number and stoichiometry of species in metal–ligand equilibria. In ESI-MS, the liquid is dispersed by its release from a positive or negative charged capillary into a strong electric field. This process produces a fine aerosol, a high number of small droplets which may carry additional charges. Afterwards, the solvent evaporates until the droplet hits its Rayleigh limit and Coulomb fission occurs.40,41 The gas phase ions are separated according to their mass to charge ratio (m/z) and finally detected, be they produced by field desorption due to the high field strength on the droplet's surface (i.e. ion evaporation model,42 IEM) or be they the result of repetitive evaporation and fission cycles (i.e. charged residue model,43 CRM). The typical time-scale of the ESI process is about 10−2 s. It is still problematic to infer correctly from ESI-MS data on the concentrations of the equilibrium species, but in several contributions the comparisons of the obtained ESI-MS data with well-established and reliable techniques are generally very good.44,45 Thus, we probed the occurrence of metal complexes in the bicarbonate solution on a tentative basis with ESI-MS. Chemical formulas, which we inferred from ESI-MS data, are deduced solely from their m/z ratio which does not contain any information of the molecular constitution. Thus the given chemical formulas should be considers as mere sum formulas; we cannot (and do not) distinguish between e.g. solvated carbon dioxide and carbonic acid (e.g. [(H2O)12(CO2)H]+vs. [(H2O)11(H2CO3)H]+). Further, we can only assume how many and which ligands are bound to the metal center; the observed/assigned species will comprise both, inner and outer coordination sphere (e.g. solvation shell), and we can neither distinguish them nor assess their protonation state. We will use in the sequel a notation that reflects the chemical situation. From our point of view, the most reliable assignments are those which belong to signal progressions.No signals appear in the positive spectrum of a saturated calcium bicarbonate solution which can be assigned to a calcium-containing species (cf. Fig. S4†). All prominent signals either appear in the case of a pure water sample or of a saturated solution of carbon dioxide. The negative ESI-MS spectrum (cf.Fig. 3), however, shows numerous signals which can be assigned to mononuclear calcium carbonate complexes and oligonuclear calcium carbonate clusters. Only one of the assignable signals is singular and cannot be assigned to a distinct progression of complexes ([Ca2(CO3)(OH)4(H2O)7]2−), whereas other signals are part of a progression which differ by 100 Dalton, i.e. the mass of one [CaCO3] unit and thus represent the stepwise growth of the complexes/clusters by one single calcium carbonate ion pair: e.g. [Ca(OH)3]− → [Ca2(CO3)(OH)3]− → [Ca3(CO3)2(OH)3]− (green set in Fig. 3), [Ca3+2n(CO3)3+2n(OH)2(H2O)7]2− (yellow set in Fig. 3 with n = 0–3) and [Ca4+n(CO3)3+n(OH)4(H2O)7]2− (n = 0–5, blue set in Fig. 3). Thus it seems reasonable to assume that only negatively charged carbonate complexes are present in solution and that their charge is the origin of the electrostatic stabilization of the emulsified state. When EDTA is added to the calcium bicarbonate solution, all species vanish in favor for two new signals: the calcium-free EDTA (290.9 Da in the negative spectrum and 293.0 Da in the positive spectrum) and the Ca–EDTA complex (328.8 Da in the negative spectrum).
![Negative ESI mass spectrum of a saturated solution of calcium bicarbonate in water. Three signal groups progress by 100 Da, which correlates to m/z of [CaCO3] and represent the stepwise growth of the clusters by one single calcium carbonate ion pair: [Can+1(CO3)n(OH)3]− (n = 0–2, green), [Ca3+2n(CO3)3+2n(OH)2(H2O)7]2− (n = 0–3, yellow) and [Ca4+n(CO3)3+n(OH)4(H2O)7]2− (n = 0–5, blue).](/image/article/2011/NR/c0nr00761g/c0nr00761g-f3.gif) | ||
| Fig. 3 Negative ESI mass spectrum of a saturated solution of calcium bicarbonate in water. Three signal groups progress by 100 Da, which correlates to m/z of [CaCO3] and represent the stepwise growth of the clusters by one single calcium carbonate ion pair: [Can+1(CO3)n(OH)3]− (n = 0–2, green), [Ca3+2n(CO3)3+2n(OH)2(H2O)7]2− (n = 0–3, yellow) and [Ca4+n(CO3)3+n(OH)4(H2O)7]2− (n = 0–5, blue). | ||
The mass spectra discussed above are not identical at all pH values; the complex formation equilibrium is pH-dependent: a saturated solution of calcium carbonate at pH = pKA2 = 10.33—which is the supernatant of an aqueous slurry of CaCO3—shows in the negative spectrum for instance signals that we can assign to hydroxo complexes, e.g. [Ca2(OH)6(H2O)12]2− at m/z = 199.0. This illustrates clearly that the formation of calcium carbonate at high pH competes with that of calcium hydroxide (KSP = 4.68 × 10−6). At pH = 9.0, none of the signals discussed above appears and the signal intensity drops considerably which may be due to the low solubility of CaCO3 at pH = 9.0. In crystallization experiments in the levitator no emulsified state could be found for solutions adjusted to pH = 9.0 and pH = 10.33. However, already after 200 s calcite crystals could be found which are assumed to be formed by an aggregation/ripening process of nanoparticles with a diameter of 20–50 nm (cf. Fig. S5†).
A measurement of the concentration of free calcium ions in the calcium bicarbonate solution with a calcium ion sensitive electrode shows it to be significantly lower than its overall concentration determined by atomic absorption spectroscopy or than a calcium chloride solution at identical calcium concentration (−3.2%). This indicates the presence of bound calcium species as reported by Gebauer et al.14,46
Based on the available experimental data it is impossible to decide whether the clusters detected by ESI-MS are fragments of the pre-nucleation clusters (fragmented due to ESI) described by Gebauer et al. or whether they can represent potential precursor species.14
Structure of equilibrium calcium complexes by XAS spectroscopy
The X-ray Absorption Near Edge Structure (XANES) of the Ca-K edge gives further evidence for the existence of complex species in the mother solution. In Fig. 4, the XANES spectrum of a calcium bicarbonate solution is compared to that of a solution of calcium chloride and that of crystalline calcite. At first glance a distinct decrease in features of the solution species compared to the crystalline calcite sample becomes obvious. Such a reduction of features typically indicates an increase in structural disorder.16 Whereas both solutions merely show one large peak at 4049 eV, crystalline calcite shows two distinct peaks at 4047 eV and 4058 eV and an additional shoulder at 4043 eV. Whereas the shoulder originates from the 1s → 4p transition, the large signals around 4049 eV arise mainly from Ca–O scattering in the first shell.47 This signal is more prominent in the calcium chloride solution than in the solution of calcium bicarbonate. Chloride is only a weakly coordinating anion, and at the chosen concentration of 7.9 mM L−1, only water is present in the first coordination shell of the calcium ions for the calcium chloride solution. One may infer that the reduction in intensity is due to water ligand exchange.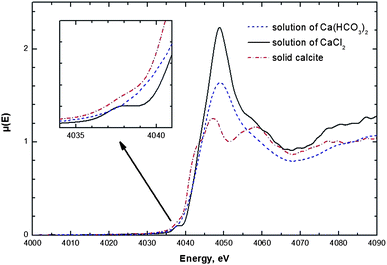 | ||
| Fig. 4 Comparison of the XANES at the Ca-K-edge of solid calcite, a solution of calcium chloride (7.9 mM L−1) and a saturated calcium bicarbonate solution (calcium concentration 7.9 mM L−1 as determined by AAS). The inset displays the pre-edge spectra which show at 4038 eV the 1s → 3d transition. This transition is formally forbidden in the case of octahedral symmetry. Only in the case of the calcium bicarbonate solution, the transition is well pronounced, in the other two cases of calcite and calcium chloride, the transition is either only weak or absent. | ||
The pre-edge range of the spectra (inset of Fig. 4) shows a well pronounced 1s → 3d transition for the calcium chloride solution. It is still present but weak for the crystalline calcite sample, whereas it is absent for the calcium bicarbonate solution. This transition is symmetry forbidden in centrosymmetric, i.e. octahedral coordination, as it seems to be the case for calcium ions in the bicarbonate solution. The transition becomes allowed by thermal disorder or quadrupolar coupling which removes the inversion symmetry even for centrosymmetrical structures such as the sixfold octahedral coordinated calcium in the calcite structure.18,47,48 Considering the presence of the pre-edge 1s → 3d transition and the small shift towards higher energy of the main signal in the case of the calcium chloride solution, it seems reasonable that fully hydrated calcium ions have a coordination number higher than six. As a result, inversion symmetry is absent and the pre-edge signal is spectroscopically allowed. This rationalization is in agreement with recent studies that propose a coordination number of seven resp. eight.47,49 This reduction of the coordination number for the calcium bicarbonate solution is likely due to the higher steric demand of carbonato ligands compared to water acting as a ligand. In summary, the XANES result indicates that the metal coordination in a calcium bicarbonate solution is highly symmetric (probably octahedral) and thus differs from that of a purely hydrated calcium species. A more elaborated and time-resolved X-ray absorption study probing the processes during emulsification is in progress.
Equilibrium Sr, Ba, Mn, Cd and Pb complex species traced by ESI-MS
The ESI mass spectra of the other metal carbonates MCO3 (M = Sr, Ba, Mn, Cd, Pb), (Fig. 5 and S6–S9†) are more complex than those of CaCO3 because they exhibit additional isotope patterns. The isotope patterns, however, allow a more reliable signal assignment than in the case of calcium bicarbonate solution because metal-free clusters can easily be distinguished from metal-centered species by their characteristic isotope patterns. The negative mass spectrum of strontium carbonate does not show extensive signal progressions as observed for calcium carbonate, but several signals are correlated by the m/z of [SrCO3]: [Sr(OH)3]− → [Sr2(CO3)(OH)3]− → [Sr3(CO3)2(OH)3]−. All other signals are singular, e.g. [Sr2(CO3)(H2O)(OH)3]−, [Sr4(CO3)(H2O)8(OH)8]2− or [Sr2(CO3)2(H2O)(OH)]−. The mass spectra of a barium bicarbonate solution show fewer signals than the spectra of the other alkaline earth bicarbonates. Interestingly, the negative mass spectrum of barium bicarbonate shows almost exclusively a stepwise ligand exchange substituting HCO3− for OH−: [Ba(OH)3]− → [Ba(HCO3)(OH)2]− → [Ba(HCO3)2(OH)]−. Lead shows a very broad isotope pattern which complicates the signal assignment severely: only few signals could be assigned, e.g. [Pb(H2O)10(H2CO3)]2+ or [Pb(OH)(CO3)]−. The signals of manganese carbonate can only be assigned, if one accounts for clusters with six or more manganese centers and hydroxo ligands, e.g. [Mn4(H2O)2(CO3)5]2−, [Mn6(OH)6(CO3)3]2−, [Mn7(CO3)7(OH)2]4− or [Mn8(CO3)7(H2O)9(OH)5]3−.![Negative ESI mass spectrum of a saturated solution of cadmium bicarbonate. Only one single peak dominates the spectrum, the spacing and the intensities of the isotope pattern indicate a single-centred, mono-charged cadmium complex: [Cd(HCO3)3(H2CO3)4(H2O)7]−.](/image/article/2011/NR/c0nr00761g/c0nr00761g-f5.gif) | ||
| Fig. 5 Negative ESI mass spectrum of a saturated solution of cadmium bicarbonate. Only one single peak dominates the spectrum, the spacing and the intensities of the isotope pattern indicate a single-centred, mono-charged cadmium complex: [Cd(HCO3)3(H2CO3)4(H2O)7]−. | ||
Only the mass spectra of the cadmium carbonate solution differ significantly from the spectra discussed above. In the positive spectrum no signals of cadmium-containing species were observed (indicated by the missing isotope pattern of cadmium). The negative spectrum, however, shows only one very prominent signal of a singly charged mononuclear cadmium complex, which can be assigned to the hydrated cluster [Cd(HCO3)3(H2CO3)4(H2O)7]− (cf.Fig. 5), i.e. it seems reasonable that only one single complex exists in solutions of cadmium bicarbonate. This cadmium complex is remarkably stable. Its stability might be attributed to a 7 + 7 (or an approximate 6 + 8, reminiscent of a bcc-like environment of the Cd2+ centre) coordination. As a result, the subsequent transformation of the amorphous phase to a crystalline state might be inhibited and thus cannot be traced by WAXS. This could be one reason for the long lifetime of the emulsional state of cadmium carbonate observed in the TEM studies.
Discussion
The occurrence of liquid precursors in six different carbonate minerals demonstrates that the non-classical crystallization route via a liquid intermediate is not a singular phenomenon of the extensively studied calcium carbonate system. It seems to be characteristic for (many) bivalent metal carbonates because the carbonate anion is the dominating agent. No additives are needed in order to stabilize or induce the liquid precursor state in the levitated droplet experiments. Our observations further provide a simple explanation why concepts applied in calcium carbonate morphosynthesis are applicable to a wider range of minerals: in all these morphogenetic processes a liquid precursor phase stabilized with the aid of polymers plays a key role (e.g. the formation of nanowires composed of earth alkaline carbonates).20,50–52It is reasonable that the occurrence of the liquid state is related to the degrees of freedom of the carbonate anions (e.g. rotational, tilting, protonation) which pose a potential barrier to crystallization. This barrier can be increased by introducing additional degrees of freedom, in the present case by the reduced symmetry of the anion and amplified by protonation. When highly interacting polymers are employed, they introduce additional degrees of freedom (besides the fact that they reduce the supersaturation by sequestering cations), which extend the lifetime of the liquid state long enough that non-equilibrium morphologies can be obtained via a PILP process, as proposed by Gower et al.30,50 Typically in such PILP processes the slow diffusion technique is employed which is based on the slow release of carbon dioxide and ammonia by thermal decomposition of ammonium carbonate which leads to an increase of pH to 9.5.37 In the absence of acidic polymers at this high pH it was shown that an emulsified state could not be traced, and a nanoparticle based process was prominent. After increasing the pH the liquid precursor was no longer observable. This effect may be compensated by addition of multidentate ligands like small acidic polymers which introduce many folds of additional interaction potentials, affect the reaction kinetics and enlarge the lifespan of the emulsion. Based on the current data, this seems to be the basis of the PILP process which leads to liquid polymer-stabilized intermediates as demonstrated by Gower et al.32,50,53
Stabilizing the liquid state by tailored acidic proteins may thus enable Nature to exploit this mineral phase in biomineralization processes of carbonate-based minerals.32 We infer that an emulsified state should be accessible for a wider range of inorganic minerals/materials beside the carbonates. One condition is that the chosen system permits a high potential of interaction between the different solute species and the solvent. This simple concept may open up a variety of potential morphosynthetic applications.30–32,50–53
In this paper, we supported our model by ESI-MS and demonstrated that mono- and multinuclear carbonate-complexes exist already in a non-supersaturated mother solution: a broad range of hydrated carbonate complexes was observed in all cases. They precede the mineral emulsion and can be assumed to be the constituents of the subsequently evolving mineral emulsion phase which obviously owes its liquid state to the high degree of hydration. The complexes traced by ESI-MS can reach a remarkable size; e.g. for calcium carbonate calcium-centred clusters up to m/z > 500 were found.§
Different protonated carbonate species coexist in a bicarbonate buffer solution at pH = 5.9–6.0, i.e. small amounts of CO32−, predominantly HCO3− (and to a small extent undissociated H2CO3).‡ These carbonate species may serve as mono- and/or bidentate ligands (e.g. η1-HCO3vs. η2-HCO3−) and eventually as bridging ligands (e.g. µ2-CO32−) for bivalent metal ions. Calcium prefers, for instance, a sixfold coordination only marginally over five- or sevenfold coordination,54 and up to ten water molecules have been reported to occur in the first hydration shell of calcium.55 Furthermore, the hydration energy of calcium is known to arise not only from the first hydration shell but to a large extent from the bulk beyond the first shell which implies that the coordination chemistry of calcium and its congeners is strongly influenced not only by inner-sphere but also by outer-sphere interactions with water or other chemical species engaged in hydrogen bonding, e.g. protonated carbonates.56 The presence of (i) the different carbonate ligands and water in the first and the second coordination sphere, (ii) the possibility for µ- and η-coordination at the metal center, (iii) the choice of coordination numbers of the metal center lead to a strong variation of the local structures which favor the formation of a strongly hydrated non-crystalline phase. As a result, the classical nucleation route is barred, and the formation of a non-classical mineral emulsion proceeds via a liquid/liquid phase separation. No crystallization nuclei will be formed whose structures closely resemble that of any crystalline mineral phases. We infer that the liquid/liquid phase separation which leads to the metal carbonate emulsions is in principle an aggregation of the detected hydrated pre-critical hydrated complexes entrapping additional solvent which may belong to the second coordination sphere. The question whether the phase separation occurs in a binodal or spinodal fashion still remains to be answered.
Except for cadmium, all clusters detected by ESI-MS seem to predominantly consist of carbonate rather than bicarbonate even though bicarbonate is the prevailing species in solution: this is most apparent in the case of the m/z progressions by single [MCO3] units, i.e. only carbonate ions appear. Our findings strongly support recent results on the existence of PNC presented by Gebauer et al. and their claim that PNCs do contain carbonate but no bicarbonate:14 the carbonate anion appears to be a stronger ligand than bicarbonate. Additionally, the hypothesis that the Ca2+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CO32− ratio in the PNC is approximately 1:1 seems to find experimental support.14
:CO32− ratio in the PNC is approximately 1:1 seems to find experimental support.14
Conclusions
In conclusion, we have shown that the homogenous formation of metal carbonates MCO3 (M = Ca, Sr, Ba, Mn, Cd, Pb) at near neutral pH proceeds via an amorphous liquid-like state which in fact behaves like a classical emulsion. This mineral emulsion is formed in the absence of any stabilizing polymers or additives at near neutral pH, and the transient mineral emulsion is stabilized electrostatically. The liquid/liquid phase separation is preceded by the presence of mono- and multinuclear carbonate complexes which already exist in the saturated mother solution. Their existence was demonstrated by ESI-MS and calcium activity measurements and supported by elementary structural information from XANES. The presented findings corroborate strongly the existence of pre-nucleation clusters in carbonate-based systems proposed by Gebauer et al.At nearly neutral conditions, weak interactions outcompete the classical nucleation of divalent metal carbonates and lead to a non-classical liquid/liquid phase separation yielding in an electrostatically stabilized mineral emulsion.
Experimental part
Saturated bicarbonate solutions were prepared by treating a slurry of the respective carbonate salt MCO3 (M = Ca, Sr, Ba, Mn, Cd or Pb; ultrapure) in ultrapure water (Millipore Synergy 185 with UV photooxidation, 18.2 MΩ cm−1) with carbon dioxide (Westfalen AG). For a couple of ESI-MS experiments, calcium carbonate (Puratronic, 99.997% on metals basis) and ultrapure water (spectrophotometric grade) from Alfa Aesar were employed. The slurry was filtered to a 20 nm cutoff with a cascade of syringe filters (0.1 µm Millipore Millex VV and 20 nm Whatman Anotop) and treated again with carbon dioxide for at least 1 h per 10 mL filtrate. The resulting bicarbonate solution is virtually free of foreign and metal carbonate nuclei as verified by SAXS and TEM in a typical experiment. The solution of calcium carbonate at pH = 10.33 was prepared by stirring a slurry of calcium carbonate (Puratronic, Alfa Aesar) in ultrapure water (spectrophotometric grade, Alfa Aesar) overnight, filtering with a cut-off of 20 nm and stirring it again for a couple of hours, diluted by at least 100 µL water per 5 mL of sample. The mother solution at pH = 9.0 was prepared by adjusting the pH of the calcium carbonate solution at pH = 10.33 with the calcium bicarbonate solution and allowing it to equilibrate for a couple of hours. The mineralization of a 4 µL droplet of the mother solution was performed contact-free by means of an ultrasonic levitator (58 kHz, Tec5, Oberursel, Germany). TEM investigations were carried out with a Phillips EM 420 running at 120 kV, equipped with an ORCA-ER Camera (1024 × 1024 pixel) and run with an AMT Image Capture Engine v5.42.540a. TEM samples were prepared by transferring a droplet to a lacey-coated TEM grid (Plano, Germany) and drying in air. A washing step was omitted because this leads to complete dissolution of the liquid transient phase.Wide-angle and small-angle X-ray scattering (WAXS, SAXS) experiments were performed at the µSpot beamline at BESSY, which provides a monochromatic beam (λ = 1.00257 Å based on calibration with corundum) and a photon flux of about 109 s−1.57 Due to a small beam diameter of 20 µm a mathematical desmearing of the experimental scattering intensity function was obsolete. Reduction of scattering data was accomplished with the data analysis program FIT2D.58
The educt solutions were characterized by mass spectrometry employing an HCT+ an Ion Trap mass spectrometer (Bruker Daltonics, Bremen) equipped with an electrospray ionization source. The measurements were done by direct infusion in positive and negative ion-mode (standard enhanced scan mode, capillary voltage: +5 kV, end plate offset −500 V, capillary exit −82.5 V, trap drive 26.1; fluxes: nebulizer pressure 35 psi (nitrogen), dry gas flow 10 L min−1 (nitrogen), dry gas temperature 365 °C). The educt solutions were used as obtained (vide supra). The assignment of the signals was accomplished employing a Python script CMCalc which was proprietary developed for this task (ActivePython v2.6.3.7 for Win32-x86, ActiveState Software Inc., Vancouver, Canada). It is based on an iterative “brute force” approach and calculates all possible combinations from a given set of constituents which meet a specified set of constraints. The script is available upon request from one of the authors (S.E.W.).
The calcium activity was estimated with the pH/ISE measuring device ProLab 3000 from Schott Instruments, employing the calcium ion selective electrode Ca1100A (Schott Instruments) and the reference electrode B2920+ (Schott Instruments).
The X-ray absorption near edge structure was probed at the undulator beamline 18 ID of the http://www.aps.anl.gov/ at Argonne National Laboratory. For details about the beamline, cf. Fischetti et al.59 All spectra were collected in fluorescence mode at room temperature. Peak position in XANES spectra was estimated via their first derivative. Data reduction was accomplished with Athena 0.8.061.60
Acknowledgements
This project was supported by the Deutsche Forschungsge-meinschaft within the priority program 1415: Kristalline Nichtgleichgewichtsphasen. We thank Simone Rolf (BAM) for technical assistance. S.E.W. thanks the Konrad Adenauer-Stiftung for a fellowship. We thank the BioCAT team at the Advanced Photon Source, especially Raul Barrea for excellent support during XAS experiments. Use of the Advanced Photon Source was supported by the US Department of Energy, Basic Energy Sciences, Office of Science, under contract no. W-31-109-ENG-38. BioCAT is a National Institutes of Health-supported Research Center RR-08630. Finally, we thank Holger Tamminga from SCHOTT Instruments GmbH for his support in estimation of the calcium activity.References
- R. Zeebe, J. Zachos, K. Caldeira and T. Tyrrell, Science, 2008, 321, 51–52 CrossRef CAS
.
- Q. Yang, Y. Liu, A. Gu, J. Ding and Z. Shen, J. Colloid Interface Sci., 2001, 240, 608–621 CrossRef CAS
- J. D. Passaretti, T. D. Young, M. J. Herman, K. S. Duane and D. Bruce, Tappi J., 1993, 76, 135–140 Search PubMed
- K. J. Snowden, K. Wu, and J. M. Rodriguez, Surface Modified Calcium Carbonate Composition and Uses Therefore, Search PubMed US Pat. 5531821, 1996.
- Y. P. Zhang, H. Shaw, H. R. Farquhar and R. Dawe, J. Pet. Sci. Eng., 2001, 29, 85–95 CrossRef CAS
-
Handbook of Biomineralization, ed. E. Bäuerlein, P. Behrens and M. Epple, Wiley-VCH Verlag GmbH, Weinheim, Germany, 2007 Search PubMed
-
H. Cölfen and M. Antonietti, Mesocrystals and Nonclassical Crystallization, Wiley-VCH Verlag GmbH, 2008 Search PubMed
- R. Becker and W. Döring, Ann. Phys., 1935, 416, 719–752 CrossRef
- M. Volmer and A. Weber, Z. Phys. Chem., 1926, 119, 277 Search PubMed
-
M. Volmer, Die chemische Reaktion, Bd. IV: Kinetik der Phasenbildung, Steinkopff, Dresden, 1939, vol. 416 Search PubMed
- H. Cölfen and M. Antonietti, Angew. Chem., Int. Ed., 2005, 44, 5576–5591 CrossRef
- M. Niederberger and H. Cölfen, Phys. Chem. Chem. Phys., 2006, 8, 3271 RSC
- N. Gehrke, N. Nassif, N. Pinna, M. Antonietti, H. S. Gupta and H. Cölfen, Chem. Mater., 2005, 17, 6514–6516 CrossRef CAS
- D. Gebauer, A. Völkel and H. Cölfen, Science, 2008, 322, 1819–1822 CrossRef CAS
- D. Gebauer, P. N. Gunawidjaja, J. Y. P. Ko, Z. Bacsik, B. Aziz, L. Liu, Y. Hu, L. Bergström, C.-W. Tai, T.-K. Sham, M. én and N. Hedin, Angew. Chem., Int. Ed., 2010, 47, 8889–8891 CrossRef
- Y. Levi-Kalisman, S. Raz, S. Weiner, L. Addadi and I. Sagi, Adv. Funct. Mater., 2002, 12, 43 CrossRef CAS
- L. Addadi, S. Raz and S. Weiner, Adv. Mater., 2003, 15, 959–970 CrossRef CAS
- R. S. Lam, J. M. Charnock, A. Lennie and F. C. Meldrum, CrystEngComm, 2007, 9, 1226 RSC
- L. Chen, Y. Shen, A. Xie, B. Huang, R. Jia, R. Guo and W. Tang, Cryst. Growth Des., 2009, 9, 743–754 CrossRef CAS
- S. J. Homeijer, R. A. Barrett and L. B. Gower, Cryst. Growth Des., 2010, 10, 1040–1052 CrossRef CAS
- I. Sondi and E. Matijević, Chem. Mater., 2003, 15, 1322–1326 CrossRef CAS
- Z. Pan, J. Tao, Y. Zhu, J. Huang and M. P. Paranthaman, Chem. Mater., 2010, 22, 149–154 CrossRef CAS
- P. van Emmerik and C. A. Smolders, Eur. Polym. J., 1973, 9, 931 CrossRef CAS
- C. A. Smolders, J. J. Aartsen and A. Steenbergen, Colloid Polym. Sci., 1971, 243, 14–20 CrossRef CAS
- V. G. Taratuta, A. Holschbach, G. M. Thurston, D. Blankschtein and G. B. Benedek, J. Phys. Chem., 1990, 94, 2140–2144 CrossRef CAS
- A. W. Perriman, H. Cölfen, R. W. Hughes, C. L. Barrie and S. Mann, Angew. Chem., Int. Ed., 2009, 48, 6242–6246 CrossRef CAS
-
(a) O. Galkin, K. Chen, R. L. Nagel, R. E. Hirsch and P. G. Vekilov, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 8479–8483 CrossRef CAS
- S. E. Wolf, J. Leiterer, M. Kappl, F. Emmerling and W. Tremel, J. Am. Chem. Soc., 2008, 130, 12342–12347 CrossRef CAS
- M. Faatz, F. Gröhn and G. Wegner, Adv. Mater., 2004, 16, 996–1000 CrossRef CAS
- L. B. Gower and D. J. Odom, J. Cryst. Growth, 2000, 210, 719–734 CrossRef CAS
- L. B. Gower and D. A. Tirell, J. Cryst. Growth, 1998, 191, 153–160 CrossRef CAS
- L. B. Gower, Chem. Rev., 2008, 108, 4551–4627 CrossRef CAS
- J. Rieger, T. Frechen, G. Cox, W. Heckmann, C. Schmidt and J. Thieme, Faraday Discuss., 2007, 136, 265 RSC
- H. Haberkorn, D. Franke, T. Frechen, W. Goesele and J. Rieger, J. Colloid Interface Sci., 2003, 259, 112–126 CrossRef CAS
- R. Tuckermann, S. Bauerecker and B. Neidhart, Phys. Unserer Zeit, 2001, 2, 69–75 CrossRef
- E. H. Trinh, Rev. Sci. Instrum., 1985, 56, 2059–2065 CrossRef CAS
-
Handbook of Chemistry and Physics, ed. D. R. Lide, CRC Press, 2002 Search PubMed
-
E. J. Verwey and J. T. Overbeek, Theory of the Stability of Lyophobic Colloids. The Interaction of Sol Particles Having a Electrical Double Layer, Elsevier Pub. Comp., Amsterdam, New York, 1948 Search PubMed
- J. B. Fenn, Angew. Chem., Int. Ed., 2003, 42, 3871–3894 CrossRef CAS
- P. Kebarle and U. H. Verkerk, Mass Spectrom. Rev., 2009, 28, 898–917 CrossRef CAS
- K. Li, H. Tu and A. K. Ray, Langmuir, 2005, 21, 3786–3794 CrossRef CAS
- J. V. Iribarne, J. Chem. Phys., 1976, 64, 2287 CrossRef CAS
- M. Dole, J. Chem. Phys., 1968, 49, 2240 CrossRef CAS
- Z. L. Cheng, K. W. Siu, R. Guevremont and S. S. Berman, J. Am. Soc. Mass Spectrom., 1992, 3, 281–288 CrossRef CAS
- V. B. Di Marco and G. G. Bombi, Mass Spectrom. Rev., 2006, 25, 347–379 CrossRef CAS
- D. Gebauer, H. Cölfen, A. Verch and M. Antonietti, Adv. Mater., 2009, 21, 435–439 CrossRef CAS
- J. L. Fulton, S. M. Heald, Y. S. Badyal and J. M. Simonson, J. Phys. Chem. A, 2003, 107, 4688–4696 CrossRef CAS
- A. Bianconi, J. Garcia and M. Benfatto, Top. Curr. Chem., 1988, 145, 26
- F. Jalilehvand, D. Spångberg, P. Lindqvist-Reis, K. Hermansson, I. Persson and M. Sandström, J. Am. Chem. Soc., 2001, 123, 431–441 CrossRef
- L. B. Gower, M. Olszta, S. Gajjeraman and M. Kaufman, Chem. Mater., 2004, 16, 2355–2362 CrossRef CAS
- M. Balz, H. A. Therese, M. Kappl, L. Nasdala, W. Hofmeister, H. Butt and W. Tremel, Langmuir, 2005, 21, 3981–3986 CrossRef CAS
- M. Balz, H. A. Therese, J. Li, J. S. Gutmann, M. Kappl, L. Nasdala, W. Hofmeister, H. Butt and W. Tremel, Adv. Funct. Mater., 2005, 15, 683–688 CrossRef CAS
- Y. Kim, L. B. Gower and E. Douglas, Langmuir, 2007, 23, 4862–4870 CrossRef CAS
- T. Ikeda, M. Boero and K. Terakura, J. Chem. Phys., 2007, 127, 074503 CrossRef
- Y. Marcus, Chem. Rev., 1988, 88, 1475–1498 CrossRef CAS
- D. Di Tommaso and N. H. de Leeuw, J. Phys. Chem. B, 2008, 112, 6965–6975 CrossRef CAS
- O. Paris, C. Li, S. Siegel, G. Weseloh, F. Emmerling, H. Riesemeier, A. Erko and P. Fratzl, J. Appl. Crystallogr., 2006, 40, 466–470
- A. P. Hammersley, S. O. Svensson, M. Hanfland, A. N. Fitch and D. Hausermann, High Pressure Res., 1996, 14, 235–248 CrossRef
- R. Fischetti, S. Stepanov, G. Rosenbaum, R. Barrea, E. Black, D. Gore, R. Heurich, E. Kondrashkina, A. J. Kropf, S. Wang, K. Zhang, T. C. Irving and G. B. Bunker, J. Synchrotron Radiat., 2004, 11, 399–405 CrossRef CAS
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS
- M. Dole, L. L. Mack, R. L. Hines, R. C. Mobley, L. D. Ferguson and M. B. Alice, J. Chem. Phys., 1968, 49, 2240–2249 CrossRef CAS
- K. Adamczyk, M. Prémont-Schwarz, D. Pines, E. Pines and E. T. Nibbering, Science, 2009, 326, 1690–1694 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available: (S1 and S5) TEM at higher magnifications and of crystallizations conducted at pH = 6.0, 9.0 and 11.3; (S2) sketch of a spreading liquid particle on a TEM grid; (S3) wide-angle scattering of BaCO3 and CdCO3; (S4 and S6–S9) ESI-MS spectra of a solution of carbon dioxide and of bicarbonates of Sr, Ba, Pb, Mn and Cd. See DOI: 10.1039/c0nr00761g |
| ‡ The amount of carbonic acid is in fact very low: at equilibrium, only a small fraction (ca. 0.2–1%) of the dissolved CO2 is actually converted to H2CO3. The first step during the formation of carbonic acid is the slow hydration step (CO2 + 2H2O → H2O + H2CO3), which is followed by a fast deprotonation (H2CO3 + H2O → H3O+ + HCO3−). It is thus widely accepted to combine these two steps yielding in the well known pKA1 = 6.3.37 Just recently, free carbonic acid was observed in real-time and a pKA1 = 3.45 was reported.62 |
| § One may further expect that the present electrospray ionization of large metal complexes proceeds via van Dole's charged residue model in which the formation and preservation of non-covalent adducts are included.39,61 |
| This journal is © The Royal Society of Chemistry 2011 |