Enhanced hydrogen storage by spillover on metal-doped carbon foam: an experimental and computational study†
George M.
Psofogiannakis
a,
Theodore A.
Steriotis
b,
Athanassios B.
Bourlinos
b,
Evangellos P.
Kouvelos
b,
Georgia Ch.
Charalambopoulou
b,
Athanassios K.
Stubos
b and
George E.
Froudakis
*a
aDepartment of Chemistry, University of Crete, PO Box 2208, Voutes, Heraklion Crete, 710 03, Greece. E-mail: frudakis@chemistry.uoc.gr; Fax: +30 2810 54500; Tel: +30 2810 545055
bNational Center for Scientific Research “Demokritos”, Agia Paraskevi Attikis, 15310, Greece. E-mail: tster@chem.demokritos.gr; Fax: +30 210 6511766; Tel: +30 210 6503614
First published on 7th January 2011
Abstract
A lightweight, oxygen-rich carbon foam was prepared and doped with Pd/Hg alloy nanoparticles. The composite revealed high H2 sorption capacity (5 wt%) at room temperature and moderate pressure (2 MPa). The results were explained on the basis of the H2 spillover mechanism using Density Functional Theory.
Storage remains a serious bottleneck for developing H2-powered vehicles. Much work has been devoted to high-surface-area solids, which are efficient stores at cryogenic temperatures1 due to weak physisorption interactions. It has also been reported that the H2storage capacity of carbon based adsorbents can be enhanced at ambient temperatures after transition metal (Pt, Pd) doping.1–6 H2 sorption on such materials has been described on the basis of the so-called “spillover” mechanism,7,8 involving H2 dissociation on the metal particles, atomic H diffusion to the surface, surface diffusion and sorption to the material's binding sites. It should also be mentioned that the very existence of spillover mechanism has been strongly debated since there are also experimental results revealing very low storage capacities after doping.9–11
Recently, we described the synthesis and characterization of a lightweight graphite-oxide-like foam possessing a rather high specific surface area (∼500 m2 g−1).12 The carbon foam (CF), derived from simple calcination of Na-chloranilate dihydrate, has the average formula C2OH and can be pictured as a porous network of interconnected bundles of turbostratically stacked graphene layers built up of aromatic and aliphatic domains with pending oxygen-containing groups (e.g.OH). CF is advantageous in the context of H2storagevia spillover as its surface is extremely active bearing oxygen containing groups, carbon-based radicals near the edges, defect-sites and radical aggregates forming spin clusters. CF has a high spin concentration (5 × 1021 spins per g),12 possibly providing an increased content of binding sites. Indeed, recent experimental studies of other groups with our CF material (inaccurately referred to as graphite oxide)7 or other oxygen rich carbons6 showed that oxygen functionalities may enhance the storage capacity of metal-doped carbons. Furthermore, since the alleged spillover process involves atomic H diffusion through the metal lattice, we considered the use of a “softer” more “malleable” alloy instead of a pure phase (e.g.Pd) in an attempt to improve kinetics, by enhancing for instance, H diffusion through crystallites. On this ground, we prepared‡ a Pd amalgam decorated composite based on CF (CF–Pd4Hg) and studied its H2storage properties.
CF is amorphous (only a broad d002 reflection12), while the composite's XRD pattern reveals reflections due to the hosted Pd4Hg particles, having the structure of potarite mineral (P4/mmm).13 The composite retains the chemical features of CF as seen by its IR spectrum (identical with that of CF12), exhibiting the characteristic carbonyl absorption at 1710 cm−1 and absorptions in the range 1500–1100 cm−1 and 600 cm−1 associated with C–OH groups. Potentiometric titrations revealed that the material has an insignificant surface acidity, implying that the –OH groups are not of phenolic nature but rather bonded to sp3 hybridized carbon atoms.
CF has a sponge-like structure (Fig. 1A) with a broad pore size distribution. The electron diffraction pattern (Fig. 1A, inset) indicates amorphous carbon and lack of long-range order. The HRTEM image of CF/Pd4Hg (Fig. 1B) shows well dispersed particles with 10 nm average size, located at the edges of graphene bundles, or embedded in the matrix with no sign of intercalation. Analysis of N2 adsorption data showed that the pore structure is maintained upon doping.
 | ||
| Fig. 1 (A) HRTEM image and ED pattern (inset) of CF. The image is inverted for clarity (carbon matrix is white). (B) HRTEM of CF/Pd4Hg. | ||
Fig. 2 shows H2 sorption isothermal gravimetric measurements (IGA, Hiden Isochema) at 298 K for CF and CF/Pd4Hg (both outgassed at ∼390 K for 48 h). CF adsorbed negligible H2 amounts (∼0.045 wt% at 20 bar) but the uptake increased remarkably after doping. Both sorption isotherms were reversible with no hysteresis in accordance with literature data.14,15 However, the CF/Pd4Hg isotherm deviates from the abundantly reported almost linear shaped ones,14,15 having a Langmuir-like profile. The total uptake is 4.6 wt% at 20 bar while, upon desorption, a residual amount (∼0.5 wt%) is retained at very low pressures. This high capacity cannot be explained solely by absorption from Pd4Hg particles (i.e.hydride formation) and indicates a synergistic effect of the nanoparticles and the support. H2 re-loading with longer equilibration times led to higher uptakes implying that the binding sites are not degenerated. It must be noted that a prolonged induction period seems necessary to initiate the process. Indeed, for the very first isothermal point (0.05 bar) of each cycle equilibration time was set to 600 min (1st run) or 1500 min (2nd run), while 15–40 min were used for all other points (Fig. 2). A 3rd cycle with 600 min induction was performed and the amount sorbed reached only 3 wt%. The pertinent kinetic curves are also shown in Fig. 2. As the same induction period was used for the 1st and 3rd run, the results point to partial deactivation of the sample presumably of “kinetic” origin. As a matter of fact for the 2nd cycle an increased induction period (1500 instead of 600 min) seems to be necessary in order to ensure equivalent uptake values for the first point of the 1st and the 2nd cycles. This kinetic deactivation is not an additive and applies only for the first cycle as shown by the identical 2nd and 3rd run kinetic curves. It is indeed anticipated that if an induction period of 1500 min was used also for the 3rd cycle, the 2nd and 3rd cycles uptake would be identical. In any case, the extended induction period coupled with the rather long equilibration times does not support the applicability of such materials in mobile H-storage systems.
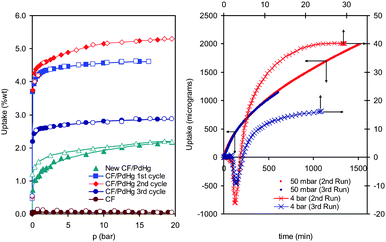 | ||
| Fig. 2 Left: H2 sorption–desorption isotherm (298 K) of the CF, original CF–Pd4Hg (3 cycles) and a 2nd CF–Pd/Hg sample (prepared from tri-hydrate). Right: kinetic uptake examples for equilibration pressures of 50 mbar and 4 bar (data from 2nd and 3rd cycle—original CF–Pd4Hg). | ||
It should be mentioned that several attempts to reproduce the sample were made. In one case the amount adsorbed was higher than the values reported above,16 however, for the majority of samples the results point to uptakes in the vicinity of 2 wt%, with signs of irreversibility (example in Fig. 2). It seems that a crucial issue and thus one of the main reasons for these discrepancies might be the use of different precursor salts. As Na-chloranilate dihydrate is no longer commercially available, new samples were prepared by either the tri-hydrate analogue or in-house prepared salts (from chloranilic acid) without controlling the number of coordinated H2O molecules. This led to varying surface areas (from 300 up to 1000 m2 g−1), but most importantly increased precursor decomposition temperatures (up to 360 °C instead of 300 °C for the dihydrate), which may destroy the sample's free radicals. In any case, the vast majority of results reveal a significant H2 sorption enhancement after doping. In this respect the reported values can be considered as a rather qualitative indication of a so-far highly debatable mechanism, highlighting mainly the importance of surface chemistry.
Aiming to further investigate the sorption mechanism we performed low pressure D2 sorption–desorption experiments coupled with mass spectroscopy (MS). A properly outgassed sample was exposed (at 298 K) to 50 mbar of D2. After equilibration the system was evacuated through a mass spectrometer (OmniStar, Pfeiffer) and the ion intensities at m/z = 2, 3, 4, 18, 19, 20 (pertaining to H2, HD, D2, H2O, HDO, D2O) were recorded vs. time. Fig. 3 presents such a plot in comparison with a blank run (no sample). As expected, the blank run is dominated by the D2 signal, which surprisingly becomes negligible upon desorption, while the lighter H2 and HD species prevail. This is direct evidence of the spillover process (but not of sorption through spillover) for two reasons: (a) D2 is dissociating and recombined with H atoms (this is the only way HD can be formed) and (b) D atoms diffuse on the carbon surface (this is the only place in our system where H is available). Additionally, a small signal from H2O also appears upon prolonged desorption and/or heating (traces of HDO were also evident) pointing to water formation upon sorption, thus raising concerns as to whether H2 is really stored or slowly (cycle after cycle) converted to water. This result might also be connected with the residual amounts of sorbate after each cycle (0.5 wt%). On these grounds the usefulness of H-storage via spillover on oxidized carbons is questioned. Nevertheless, our experimental results constitute a framework that deserves further theoretical and experimental investigation rather than rejection of the concept before all of its aspects have been thoroughly studied.
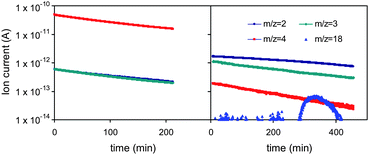 | ||
| Fig. 3 D2 desorption-MS data (298 K). Left: blank (no sample). Right: desorption from CF–Pd4Hg. | ||
Seeking to gain insight into spillover energetics, Density Functional Theory (DFT) calculations for the exemplified case of a Pd-doped oxidized graphitic surface were performed with the Turbomole program, using the B-P86 functional and the def2-TZVPP basis sets, as well as the corresponding effective core potentials for Pd, described elsewhere.8 The aim of the computations is to show that the elementary reactions involved in the spillover process (H2 dissociation, H migration to oxidized graphene, and H diffusion on oxidized graphene) are kinetically efficient. It is known that at room temperature metallic Pd dissociates molecular H2. We performed a full geometry optimization for a Pd4 cluster surrounded by 7 H2 molecules originally placed 3.5–4.0 Å away from the preoptimized cluster (Fig. 4, left).
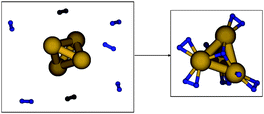 | ||
| Fig. 4 Initial and final states of DFT geometry optimization for the adsorption and dissociation of H2 on a Pd4 cluster. | ||
In the final optimized structure complete dissociation of one H2 molecule was identified, while the other 6 H2 molecules remain molecularly adsorbed in a precursor state with elongated H–H bond length. The results suggest that atomic H forms when H2 contacts Pd nanoparticles. The strongest binding for a H atom on the Pd4 cluster is at a 3-fold site with 3.2 eV binding strength, while 1-fold coordinated H was found to have a strength of 2.8 eV versus free H. The binding strength of H on Pd(111) was calculated 2.88 eV.17 Although the possible positive effect of the amalgam, compared to pure Pd, has not yet been investigated by DFT, we can speculate that Hg may assist in the migration of atomic H from the nanoparticle to the surface C atoms, possibly providing an energetically favorable path.
Pd presence triggers the generation of atomic H (or D). However, it cannot explain either the recombination to HD or the high capacity, requiring the migration of atomic H from the nanoparticle to the material. The metal-phase essentially provides kinetically efficient dissociative and associative pathways for the H atoms to adopt the pressure-dependent thermodynamic partitioning between the molecular gaseous and atomic adsorbed states. Thus, possible binding sites for atomic H on the oxidised CF were identified through DFT.
In general the oxidized graphene planes of graphite oxide surfaces are mainly populated by –OH groups as well as epoxide (C–O–C bridges) while carbonyl groups (such as C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) can appear on the periphery of graphite layers.18 CF has the average composition C2OH, and the most abundant surface species is likely to be –OH, while CO groups have been also identified. The calculations showed that atomic H binds with 3.7 eV on CO and 3.1 eV on C–O–C (the models used are shown in Fig. 5). The high binding strengths indicate that atomic H can migrate from Pd to the surrounding O functionalities, as this migration step is downhill in energy. Nevertheless, the high H-capacities measured on doped CF cannot be justified if only O atoms can accept the spillover H.
O) can appear on the periphery of graphite layers.18 CF has the average composition C2OH, and the most abundant surface species is likely to be –OH, while CO groups have been also identified. The calculations showed that atomic H binds with 3.7 eV on CO and 3.1 eV on C–O–C (the models used are shown in Fig. 5). The high binding strengths indicate that atomic H can migrate from Pd to the surrounding O functionalities, as this migration step is downhill in energy. Nevertheless, the high H-capacities measured on doped CF cannot be justified if only O atoms can accept the spillover H.
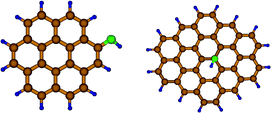 | ||
| Fig. 5 Final states of DFT geometry optimization for adsorption of H on a carbonyl O on graphite (left) and an epoxide O (right). | ||
DFT calculations showed that C atoms connected with C–OH carbons can strongly bind H (Fig. 6). The ortho configuration of H and OH on the same ring confers additional stability, just like H-pairing on graphene at ortho sites, previously explained through breaking of the radical state induced by the first adsorbed species.19 The binding strength of H bound ortho to OH was calculated 2.45 eV, in contrast to H binding on a pristine graphene plane with only 0.7 eV. The 2.45 eV binding energy is only slightly lower than the H binding strength on Pd. As a result H can probably migrate from the Pd nanoparticles to the surface C atoms adjacent to OH groups.
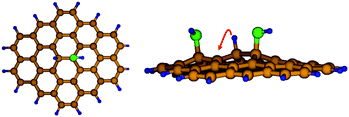 | ||
| Fig. 6 Left: optimized structure of H and OH bound to graphite. Right: migration of H on C atoms of oxidized graphite. | ||
It is anticipated that H atoms can also diffuse on the surface, as the high degree of oxidation provides a continuous network of strong binding sites. Fig. 6 shows the initial state of an Internal Reaction Coordinate calculation for the migration of an atomic H between C-atoms that are both ortho to OH group. The energy barrier for this elementary step was found ∼1.0 eV, showing that migration is slow, in agreement with the sorption experiments, but by no means impossible at room temperature. The binding of H on C atoms adjacent to OH groups can explain the enhanced capacity of the oxidised CF. The large population of functionalities on CF and the high dispersion of nanoparticles will increase the effectiveness of the H migration process. However, a fraction of these functionalities, particularly hydroxyl groups, can be irreversibly lost as water during the spillover process. Thus, the average composition of the oxidized foam will be modified by H spillover.
The thermodynamic propensity for water formation during H spillover, observed in the D2 sorption/MS experiments, was also studied via DFT. It was found that H2O formation through the reaction graphite–OH + Hfree → graphite–H2O is exothermic by −4.9 eV. Furthermore, H2O production by recombination of the H–OH pair was also found exothermic by −2.46 eV, verifying that water may be formed from pending OH groups. The additional stability of –O or –OH groups in larger structures (i.e. OH-structures or O line defects) may partially compensate the tendency to produce water. Nevertheless, the computations show that –OH surface groups not only assist the spillover of atomic hydrogen and thus enhance the uptake but also cause hydrogen loss by water formation. Irreversible formation of water during H2 charging–discharging cycles could be detrimental to the H2 capacity of CF material and deserves further investigation.
Financial support for G.P. from a Marie-Curie IRG grant and partial funding by the EC project NESSHY (FP6 contract SES6-518271) are acknowledged.
Notes and references
- A. Lueking and R. T. Yang, AIChE J., 2003, 49, 1556 CrossRef CAS.
- A. J. Lachawiec, S. G. Qi and R. T. Yang, Langmuir, 2005, 21, 11418 CrossRef CAS.
- A. Lueking and R. T. Yang, Appl. Catal., A, 2004, 265, 259 CrossRef CAS.
- Y. W. Li and R. T. Yang, J. Am. Chem. Soc., 2006, 128, 726 CrossRef CAS.
- Q. Li and A. D. Lueking, Prepr. Pap. - Am. Chem. Soc., Div. Fuel Chem., 2008, 53, 1 Search PubMed.
- L. Wang, F. H. Yang, R. T. Yang and M. A. Miller, Ind. Eng. Chem. Res., 2009, 48, 2920 CrossRef CAS.
- L. Chen, A. C. Cooper, G. P. Pez and H. Cheng, J. Phys. Chem. C, 2007, 111, 18995 CrossRef CAS.
- G. M. Psofogiannakis and G. E. Froudakis., J. Phys. Chem. C, 2009, 113, 14908 CrossRef CAS.
- C. I. Contescu, C. M. Brown, Y. Liu, V. V. Bhat and N. C. Gallego, J. Phys. Chem. C, 2009, 113, 5886 CrossRef CAS.
- N. P. Stadie, J. J. Purewal, C. C. Ahn and B. Fultz, Langmuir, 2010, 26, 15481 CrossRef CAS.
- R. Campesi, F. Cuevas, M. Latroche and M. Hirscher, Phys. Chem. Chem. Phys., 2010, 12, 10457 RSC.
- A. Bourlinos, Th. Steriotis, M. Karakassides, Y. Sanakis, V. Tzitzios, C. Trapalis, E. Kouvelos and A. Stubos, Carbon, 2007, 45, 852 CrossRef CAS.
- K. Terada and F. W. Cagle, Am. Mineral., 1960, 45, 1093 CAS.
- S. Zacharia, S. Rather, S. W. Hwang and K. S. Nahm, Chem. Phys. Lett., 2007, 434, 286 CrossRef CAS.
- Y. Li and R. T. Yang, J. Am. Chem. Soc., 2006, 128, 8136 CrossRef CAS.
- M. Miller and R. Page, DOE Annual Merit Review Meeting, Washington, DC, June 9–13, 2008 Search PubMed.
- J. Greeley and M. Mavrikakis, Nat. Mater., 2004, 3, 810 CrossRef CAS.
- W. Cai, R. D. Piner, F. J. Stadermann, S. Park, M. A. Shaibat, Y. Ishii, D. Yang, A. Velamakanni, S. J. An, M. Stoller, J. An, D. Chen and R. S. Ruoff, Science, 2008, 321, 1815 CrossRef CAS.
- Y. Ferro, D. Teillet-Billy, N. Rougeau, V. Sidis, S. Morisset and A. Allouche, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 085417 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Characterisation and experimental details. See DOI: 10.1039/c0nr00767f |
‡ Material synthesis: CF synthesis has been described elsewhere.12 CF was suspended in de-ionized water, followed by dissolution of anhydrous PdCl2 and HgCl2 (Panreac), while NaBH4 was used as a reducing agent. The suspension was centrifuged, washed thoroughly with water/acetone, and dried (65 °C) affording a black solid with a total metal content of ∼12 wt% and a molar ratio (Pd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Hg) of 4. :Hg) of 4. |
| This journal is © The Royal Society of Chemistry 2011 |