Catalytic activity of gold supported on ZnO tetrapods for the preferential oxidation of carbon monoxide under hydrogen rich conditions†
Eva
Castillejos
a,
Revathi
Bacsa
a,
Antonio
Guerrero-Ruiz
b,
Inmaculada
Rodríguez-Ramos
b,
Lucien
Datas
c and
Philippe
Serp
*a
aLaboratoire de Chimie de Coordination UPR CNRS 8241 Composante ENSIACET, ToulouseUniversity, 4, Allée Monso, 31030, Toulouse, Cedex 4, France. E-mail: philippe.serp@ensiacet.fr; Fax: +33534323596; Tel: +33534323572
bInstituto de Catálisis y Petroleoquímica, Unidad Asociada “Group for Design Appl. Het. Catal.” UNED-CSIC, C/Marie Curie 2, Cantoblanco, 28049, Madrid, Spain
cCNRS, Institut Carnot Cirimat, F-31062, Toulouse, France
First published on 23rd December 2010
Abstract
It is reported that 3 nm gold nanoparticles deposited on ZnO tetrapods show high activity for the selective oxidation of carbon monoxide in hydrogen rich streams; the catalytic activity of this system is at least twice as high as the values hither to observed on any conventional support for this reaction.
Hydrogen containing gas mixtures for polymer electrolyte membrane fuel-cells (PEMFCs) should be free of CO to avoid poisoning of the Pt-based catalyst. Presently, the hydrogen gas is produced by steam reforming of light hydrocarbons, a process which produces a mixture of gases that also contains (1–3%) CO.1 Even very small amounts of CO can poison a pure platinum electrocatalyst. Preferential oxidation of CO (CO-PROX) has been recognized as one of the most direct and productive methods to achieve acceptable CO concentrations, typically below 100 ppm.2Purification of CO containing hydrogen is complicated by the simultaneous oxidation of both H2 and CO. In addition, CO can also react with hydrogen, and unless the CO concentration in the reactant stream is sufficiently low, this reaction can also consume relatively large amounts of hydrogen, particularly at high temperatures. In order to develop PEMFC for practical applications, it is therefore necessary to design catalysts that can be used at low temperatures to remove CO selectively in a H2-rich system by oxidizing it into CO2, while at the same time being inactive for the oxidation of H2. Factors such as the choice of the support, the catalyst particle size and the deposition method are expected to play a key role in the design of such catalytic system. Nanoparticles of gold supported on oxides have been found to be particularly active for this reaction.3–7 In this context, nanocrystalline fine powders of ZnO are ideal candidates for gold deposition, mainly due the presence of acid (Zn2+) and basic (O2−) surface sites and the possibility of electron transfer from the adsorbent to the adsorbate or vice versa.8,9 In addition, they are easy to prepare and are cost effective when compared to rare earth oxide supports. Recently, we have reported the synthesis of Au/ZnO nanocatalysts and their application as efficient catalysts for CO oxidation at high temperatures in the absence of hydrogen.10 In the present communication, we describe the remarkable catalytic activity of very small gold nanoparticles deposited on ZnO tetrapods (ZnOT) for the preferential oxidation of carbon monoxide in hydrogen rich streams, for which 100% conversion has been achieved at temperatures lower than 360 K. Our experimental observations show that the unique surface structure of the tetrapods11–15 allows to efficiently control both metal–support interaction and the size distribution of Au particles when compared to commercially available nanocrystalline ZnO (ZnOD) with a similar specific surface area.
The ZnO tetrapods henceforth denoted by ZnOT were synthesized by chemical vapour synthesis, the details of which are given in previous publications.16,17 The synthesis of ZnO tetrapods, Au/ZnO catalysts and their characterization, as well as experimental aspects concerning the PROX reaction are given as ESI (Fig. S1†).
For all the supports investigated, X-ray diffractograms showed the presence of highly crystalline ZnO wurtzite (JCPDS 36-1451) phase without any preferred orientation. Lattice parameter measurements did not present any deviation from standard values showing that the ZnO supports were stoichiometric. Raman spectra of both supports showed the characteristic peaks due to wurtzite type structure and the strong peak at 438 cm−1 corresponding to the high energy non-polar E2 (high) mode involving the oxygen sublattice. The narrow line shape (FWHM = 6 cm−1) indicated in both cases a high crystalline order. For ZnOT, SEM images confirmed the presence of tetrapods formed from nanorods of diameters in the range 8–40 nm and lengths from 100 nm to 1 µm giving them an aspect ratio of around 100 (Fig. 1a). High resolution TEM images showed that the rods were single crystals (see Fig. S2†). BET surface areas calculated from N2 adsorption isotherms (type IV isotherms were observed) gave a value ranging from 18–22 m2 g−1. The total pore volume was in the range 0.06 cm3 g−1. ZnOD consists of nanocrystals of different shapes with sizes in the 20–50 nm range (Fig. 1b). A BET surface area of 20 m2 g−1 and a pore volume of 0.1 cm3 g−1 were measured.
 | ||
| Fig. 1 SEM images of zinc oxide supports (a) laboratory-grown tetrapods ZnOT and (b) commercial ZnOD. | ||
Fig. 2 shows TEM images of Au nanoparticles (NPs) supported on ZnO supports. Generally, most of the ZnO tetrapods are not damaged during the gold deposition procedure, and only few of them have been broken into individual rods. Au1/ZnOT catalyst (Fig. 2a and b) shows finely divided and highly monodisperse gold particles deposited on the support with an average size of 1–2 nm. Most of the particles are equiaxed and decorate the ZnO rods uniformly along their length. Some of the particles, especially at the edges of the tetrapods, show elongated shapes too. The Au2/ZnOT sample (not shown) shows slightly larger particles that are also homogeneously distributed on the support. The Au/ZnOD shows larger particles with a wider distribution (Fig. 2c and d). The particle size histograms are shown in Fig. 3; statistical average sizes and the Au loading are reported in Table 1. Higher metal loadings were achieved on the ZnOT support for both NaOH and Na2CO3 pointing to a different interaction between the gold precursor HAuCl4 and the two ZnO supports.
 | ||
| Fig. 2 TEM images of (a) and (b) Au1/ZnOT; (c) and (d) Au/ZnOD. | ||
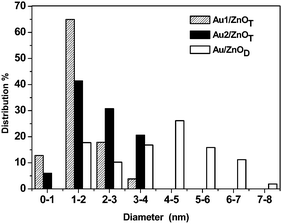 | ||
| Fig. 3 Particle size distributions determined from TEM for Au1/ZnOT, Au2/ZnOT and Au/ZnOD. | ||
Looking at the shape of the gold NPs, a difference was noticed between the two supports. On ZnOT, the NPs are relatively flat (hemispherical NPs) suggesting a strong interaction with the support, whereas on ZnOD, the gold NPs are more spherical. Interestingly, it has been observed that generally, the TOF for CO oxidation over hemispherical NPs are much higher compared to spherical particles.18
Fig. 4 shows a high resolution TEM image of a gold particle epitaxially deposited on a ZnO tetrapod. The particle is a single crystal with its (111) plane parallel to the (0002) plane of ZnO. The d value for Au (111) is 0.235 nm and that for ZnO (0002) is 0.26 nm, which shows a mismatch greater than 10%. Measurements of the interplanar distances show that the gold crystal expands significantly in the interface region to enable epitaxial growth. Further from the zone of epitaxy, the interplanar distances are closer to the values observed for pure gold. For larger gold particles twinning of the crystal is observed, presumably to relieve the strain arising from the lattice mismatch, but in very small particles, the surface and the inner atoms appear to have greater freedom of rearrangement to accommodate differences in lattice parameters. Several particles were observed and roughly around 20% of the particles were found to have epitaxial relation with the ZnOT crystal. On the other hand, no epitaxy was observed for any of the particles deposited on ZnOD. In an earlier work, we have shown that the optical properties of ZnO are shape dependent.16 We have therefore studied the optical properties, namely, diffuse reflectance and luminescence spectra of the two supports (see Fig. S3 and S4†). Luminescence spectra are particularly informative since the presence of defects is known to introduce new emission bands in ZnO, whereas perfect ZnO crystals emit predominantly in the UV.19 Diffuse reflectance measurements on the two powders show the presence of an extended absorption tail extending into the visible region for ZnOD, whereas ZnOT shows only band gap absorption. Correspondingly, luminescence spectra showed the presence of both the UV and the green emission bands for the ZnOD, whereas the tetrapods showed only the UV emission. The origin of the green band in ZnO is controversial but it is generally associated with defects and in this case, it appears that these are present on the surface as indicated by the diffuse reflectance from the two powder surfaces. This result points to the presence of surface defects in the ZnOD that could be responsible for the absence of epitaxy. Indeed, early studies on gold epitaxy on ZnO crystals have shown the need for defect free crystals to ensure epitaxial growth of gold.20
 | ||
| Fig. 4 High resolution electron microscopy image of epitaxially grown gold nanoparticles on ZnOT. | ||
X-Ray diffractograms of the Au/ZnO samples (see Fig. S5†) showed the presence of the ZnO wurtzite structure for all the three catalysts. No peaks due to gold could be detected for Au1/ZnOT and Au2/ZnOT, presumably due to the ultrafine particle sizes and low metal loading. For Au/ZnOD, which present a higher Au particle size, a very small peak corresponding to Au(111) is detected at 2θ = 38.4°. Surface semiquantitative (XPS) analyses after and before catalysis showed binding energies at 83.6 eV for the Au4f7/2 that are typical values for Au(0)21 confirming the formation of metallic Au NPs on the ZnO surface (see Fig. S6†). The Au4f5/2 peak could not be seen because of an overlap with the Zn3p3/2 peak.
The support and catalyst characterization results show that, under similar experimental conditions, the ZnO tetrapod support enables gold deposition with a higher loading and a narrower particle size distribution. Since no –OH groups were detected by infrared spectroscopy on the pristine supports this result may be due to a higher degree of functionalisation of the ZnOT surface under the conditions of Au deposition that leads to a higher nucleation rate for gold crystals. Since the particle density is higher when NaOH is used (lower sizes for the same metal loading), when compared to Na2CO3 it can be inferred that the presence of –OH groups on the tetrapod surface is responsible for the creation of these nucleating sites, and not other defects since we have shown that the ZnOD support presents a higher concentration of surface defects without resulting in higher gold loadings.
We have compared the catalytic systems, Au1/ZnOT, Au2/ZnOT, and Au/ZnOD for the CO-PROX in a H2-rich stream (PROX). Fig. 5 shows the CO oxidation performances of these systems. At 360 K these catalysts give a catalytic activity (Table 1) of 1.65, 4.06 and 2.43 molCOgAu−1 h−1, respectively, and no catalyst deactivation is observed during the 50 h run. The reaction performances of Au2/ZnOT catalyst with time on stream are shown in Fig. S7†.
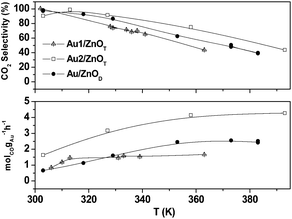 | ||
| Fig. 5 CO conversion and selectivity towards CO2vs. temperature. | ||
The Au2/ZnOT exhibits the highest CO conversion and selectivity in the temperature range investigated, the conversion reaching 100% at 360 K. This activity for CO conversion is twice as high as that reported for a Au/ZnO nanocatalyst for high temperature CO oxidation in the absence of hydrogen.10 It has been generally observed7 that if Au catalysts can selectively oxidize CO to CO2 in the presence of H2, when the reaction temperature is increased the selectivity is affected. The Au2/ZnOT catalyst shows the higher selectivity for CO oxidation in the presence of H2, with values close to 100% at 310 K and 65% at 370 K. This catalyst does not show any decrease of its performances after five consecutive CO-PROX cycles of 10 h each. The CO conversion achieved under CO-PROX mixtures for the Au2/ZnOT sample is remarkably high compared to the values reported in the literature for gold catalysts on any conventional support (Table 2). In order to get a more accurate comparison between the three catalysts tested we also calculated the activity per surface atom of gold (Table 1). Indeed, it has been suggested that for small gold nanoparticles the whole reaction takes place on the gold surface,22 in contrast to the Bond and Thompson mechanism involving oxidized gold species at the gold–support interface.23 In our case, the role of the support would be mainly to stabilize the small gold particles. The calculated activities were 2.3, 8.1 and 8.8 molCO gAu-surf.−1 h−1, for Au1/ZnOT, Au2/ZnOT, and Au/ZnOD, respectively. The better performance of Au2/ZnOT compared to Au/ZnOD is attributed to a better dispersion of gold on the ZnOT support. It has been well documented that nanoparticles of gold with less than 10 nm size show increased catalytic activity especially at low temperatures.18 It is not clear, however, why there is a difference in activity between the Au1/ZnOT and the Au2/ZnOT catalysts. We propose that this difference might arise from the presence of a large number of sub-nanometre sized particles in Au1/ZnOT and that there may be an optimum size (3–5 nm) to achieve high activity.3,21 It has been shown that gold particles below ∼2.5 nm are significantly less active than particles of 3 nm for CO electro-oxidation,25 and that small gold clusters are deactivated by gas phase oxygen at high temperatures.26 This difference of reactivity of sub-nanometre sized particles may also arise from the epitaxy of gold. Our HRTEM results show that for 2.2 nm epitaxially grown particles, the interplanar distances can be changed by 10% at the interface leading to strain in the particles. For smaller particles as the ones present in Au1/ZnOT, we can assume that the strain will be more pronounced for the surface atoms compared to Au2/ZnOT. On their observations of aberration free phase image of a gold particle on TiO2, Kawasaki et al. concluded that fine gold particles are indeed deformed more easily than larger sized particles.27 Early theoretical studies by Mavrikakis et al. have shown a general correlation between the surface strain and energies for gas adsorption on metal particles.28 Thus, if for some reactions such atomic rearrangement may have a positive role on the catalytic activity of Au NPs, in our case it could be the opposite.
In conclusion, we have reported the higher catalytic performances of gold nanoparticles supported over ZnO tetrapods in the preferential oxidation of CO in hydrogen rich streams when compared to any commercial support with a similar porosity. This study illustrates the effect of substrate nanostructure on the catalytic activity of the Au/ZnO system and can have important implications for the production of new catalytic systems presenting high selectivity.
Acknowledgements
E.C. gratefully acknowledges financial support from the Spanish Ministry of Science and Technology and the Institute of Catalysis and Petroleochemistry (Spain). The authors thank D. Neumeyer for BET surface area measurements and S. Le Blond du Plouy (TEMSCAN, Toulouse University) for the SEM images. This work was partly supported by the Spanish Government (contract CTQ-2008-06839-C03-01/PPQ and CTQ-2008-CO3-03/PPQ).References
- M. Rydén and A. Lyngfelt, Int. J. Hydrogen Energy, 2006, 31, 1271 CrossRef CAS.
- E.-Y. Ko, E. D. Park, H. C. Lee, D. Lee and S. Kim, Angew. Chem., Int. Ed., 2007, 46, 734 CrossRef CAS.
- S. Carrettin, P. Concepcion, A. Corma, J. M. Lopez and V. F. Puntes, Angew. Chem., Int. Ed, 2004, 43, 2538 CrossRef CAS.
- O. H. Laguna, F. Romero Sarria, M. A. Centeno and J. A. Odriozola, J. Catal., 2010, 276, 360 CrossRef CAS.
- S. Ivanova, V. Pitchon, C. Petit and V. Caps, ChemCatChem, 2010, 2, 556 Search PubMed.
- P. Naknam, A. Luengnaruemitchai and S. Wongkasemjit, Int. J. Hydrogen. Energy, 2009, 34, 9838 CrossRef CAS.
- C. Louis in Nanoparticles and Catalysis, ed. D. Astruc, Wiley-VCH, Weinheim, 2008, ch. 15, pp. 475–503 Search PubMed.
- K. Sai Krishna, G. Vivekanandan, D. Ravinder and M. Eswaramoorthy, Chem. Commun., 2010, 46, 2989 RSC.
- D. Thompson, Gold Bull., 1999, 32, 12 CAS.
- S. A. C. Carabineiro, B. F. Machado, R. Bacsa, P. Serp, G. Dražic, J. L. Faria and J. L. Figueiredo, J. Catal., 2010, 273, 191 CrossRef CAS.
- L. Manna, D. Milliron, A. Meisel, E. C. Scher and A. P. Alivisatos, Nat. Mater., 2003, 2, 382 CrossRef CAS.
- M. C. Newton and P. A. Warburton, Mater. Today, 2007, 10, 50–54 CrossRef CAS.
- U. Ozgur, Y. I. Alivov, C. Liu, A. Teke, M. A. Reshchikov, S. Dogan, V. Avrutin, S.-J. Cho and H. Morkoc, J. Appl. Phys., 2005, 98, 041301 CrossRef.
- A. M. Mohammad, A. I. Abdelrahman, M. S. El-Deab, T. Okajima and T. Ohsaka, Colloids Surf., A, 2008, 318, 78 CrossRef CAS.
- Z. L. Wang, J. Phys.: Condens. Matter, 2004, 16, 829.
- R. Bacsa, J. Dexpert-Ghys, M. Verelst, A. Falqui, B. Machado, W. S. Bacsa, P. Chen, S. M. Zakeeruddin, M. Graetzel and P. Serp, Adv. Funct. Mater., 2009, 19, 875 CrossRef CAS.
- N. Reuge, R. Bacsa, P. Serp and B. Caussat, J. Phys. Chem. C, 2009, 113, 19845 CrossRef CAS.
- M. Haruta, Chem. Rec., 2003, 3, 75 CrossRef.
- A. Djurisic, W. H. Choy, V. Roy, Y. Leung, C. Kwong, K. Cheah, T. Gundu Rao, W. Chan, H. Fei Lui and C. Surya, Adv. Func. Mater., 2004, 14, 856 CrossRef CAS.
- E. F. Wasserman and K. A. Polacek, Surf. Sci., 1971, 28, 77 CrossRef.
- Z.-M. Qi, H.-S. Zhou, N. Matsuda, I. Honma, K. Shimada, A. Takatsu and K. Kato, J. Phys. Chem. B, 2004, 108, 7006 CrossRef CAS.
- M. Valden, X. Lai and D. W. Goodman, Science, 1998, 281, 1647 CrossRef CAS.
- C. G. Bond and D. T. Thompson, Gold Bull., 2000, 33, 41 CAS.
- A. Hornes, A. B. Hungria, P. Bera, A. López-Cámara, M. Fernandez-Garcia, A. Martínez-Arias, L. Barrio, M. Estrella, G. Zhou, J. J. Fonseca, J. C. Hanson and J. A. Rodríguez, J. Am. Chem. Soc., 2010, 132, 34 CrossRef CAS.
- B. E. Hayden, D. Pletcher, M. E. Rendall and J.-P. Suchsland, J. Phys. Chem. C, 2007, 111, 17044 CrossRef CAS.
- A. N. Pestryakov, N. Bogdanchikova, A. Simakov, I. Tuzovskaya, F. Jentoft, M. Farias and A. Díaz, Surf. Sci., 2007, 601, 3792 CrossRef CAS.
- T. Kawasaki, Y. Takai and R. Shimizu, Appl. Phys. Lett., 2001, 79, 3509 CrossRef CAS.
- M. Mavrikakis, B. Hammer and J. K. Norskov, Phys. Rev. Lett., 1998, 81, 2819 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental (Fig. S1); an HRTEM image of a single nanorod (Fig. S2); the diffuse reflectance spectra and luminescence spectra of ZnOT and ZnOD respectively (Fig. S3 and S4); and the X-ray diffractograms of AuZnOT, Au2ZnOT and AuZnOD (Fig. S5). See DOI: 10.1039/c0nr00724b |
| This journal is © The Royal Society of Chemistry 2011 |