Incorporating functionalized polyethylene glycol lipids into reprecipitated conjugated polymer nanoparticles for bioconjugation and targeted labeling of cells†
Prakash K.
Kandel
,
Lawrence P.
Fernando
,
P. Christine
Ackroyd
and
Kenneth A.
Christensen
*
Department of Chemistry, Clemson University, Clemson, SC 29634, USA. E-mail: kchris@clemson.edu; Fax: +1 864 656 0567; Tel: +1 864 656 0930
First published on 13th December 2010
Abstract
We report a simple and rapid method to prepare extremely bright, functionalized, stable, and biocompatible conjugated polymer nanoparticles incorporating functionalized polyethylene glycol (PEG) lipids by reprecipitation. These nanoparticles retain the fundamental spectroscopic properties of conjugated polymer nanoparticles prepared without PEG lipid, but demonstrate greater hydrophilicity and quantum yield compared to unmodified conjugated polymer nanoparticles. The sizes of these nanoparticles, as determined by TEM, were 21–26 nm. Notably, these nanoparticles were prepared with several PEG lipid functional end groups, including biotin and carboxy moieties that can be easily conjugated to biomolecules. We have demonstrated the availability of these end groups for functionalization using the interaction of biotin PEG lipid conjugated polymer nanoparticles with streptavidin. Biotinylated PEG lipid conjugated polymer nanoparticles bound streptavidin-linked magnetic beads, while carboxy and methoxy PEG lipid modified nanoparticles did not. Similarly, biotinylated PEG lipid conjugated polymer nanoparticles bound streptavidin-coated glass slides and could be visualized as diffraction-limited spots, while nanoparticles without PEG lipid or with non-biotin PEG lipid end groups were not bound. To demonstrate that nanoparticle functionalization could be used for targeted labelling of specific cellular proteins, biotinylated PEG lipid conjugated polymer nanoparticles were bound to biotinylated anti-CD16/32 antibodies on J774A.1 cell surface receptors, using streptavidin as a linker. This work represents the first demonstration of targeted delivery of conjugated polymer nanoparticles and demonstrates the utility of these new nanoparticles for fluorescence based imaging and sensing.
Introduction
The use of highly fluorescent nanoparticles as labels for cellular imaging and in vitro assays is an extremely promising approach to maximize sensitivity and minimize the limit of detection. Such nanoparticles include inorganic semiconductor quantum dots (QDs),1,2dye-doped silica particles,3 and commercially available dye-loaded latex spheres. These nanoparticles offer numerous advantages over traditional organic dyes, including bright fluorescence and improved photostability. As a consequence, great efforts have been invested in preparation of highly fluorescent nanoparticles and their use in a wide variety of applications that include biosensing, live cell imaging, and intracellular dynamics.4–6 However, use of existing nanoparticles is not without disadvantages. For example, limited dye loading due to self-quenching and undesirable leakage of small dye molecules has been reported for dye-doped silica nanoparticles3 and cytotoxicity due to leached metal from the nanocrystal core is a critical problem for use of QDs.7–9 While heavy metal leaching has been reduced by coating QDs with a variety of materials, such coatings can have their own associated cytotoxic effects7,10 and may not completely ameliorate heavy metal leakage.The limitations of current fluorescent nanoparticles provide impetus for the design of new nanoparticles with high photostability and bright fluorescence, but with greatly reduced cytotoxicity. One promising strategy is the development of conjugated polymer nanoparticles (CPNs). These nanoparticles are formed by collapse of highly fluorescent conjugated hydrophobic polymers to form nanoparticles with high absorption cross-sections and high radiative rates.11,12 The result is extraordinarily bright fluorescent nanoparticles. Because these CPNs are composed of relatively benign constituents with intrinsic fluorescence, they have low cytotoxicity,13 and cannot leach dye or constituent materials. As a result, CPNs have established themselves as useful optical probes that can be used at extremely low concentrations. However, the extreme hydrophobicity of CPNs leads to aggregation at high concentrations, thus limiting the amount of CPNs that can be added to cells in culture. In addition, this category of nanoparticle has not previously been conjugated to useful biomolecules for targeted delivery to cells.
One approach to reduce the hydrophobicity of CPNs would be to introduce hydrophilic functional group(s) to the conjugated polymer starting material(s). However, this approach could alter the structure of the polymer and affect relevant optical properties. Another strategy is to envelope the CPNs with hydrophilic component(s), which would alter the nanoparticle surface characteristics without changing polymer optical qualities.14,15 We were intrigued by reports that polyethylene glycol (PEG) with an attached phospholipid (PEG lipid) has been used to provide hydrophilicity to an otherwise hydrophobic nanosensor,16 to polymer coated quantum dots17–20 and to semiconductor polymer nanospheres formed by miniemulsion and also referred to as semiconductor polymer nanospheres.21,22 We speculated that a similar strategy could be used with CPNs formed by reprecipitation. As PEG lipids are commercially available and PEG has been widely used in biological systems, surface modification of CPNs with functionalized PEG lipids is a viable method to create more hydrophilic nanoparticles. Importantly, PEG lipids can be functionalized with a variety of end groups to incorporate a moiety for linking biomolecular recognition elements to the CPN surface. As a result, functionalized PEG lipids not only improve the hydrophilicity and biocompatibility of CPNs for live cell imaging, but also allow specific labeling of cellular targets.
Here we report a general method that uses the straightforward reprecipitation method to prepare highly fluorescent CPNs that incorporate functionalized PEG lipids, using commercially available materials. The result is functionalized soluble nanoparticles of small size that are highly stable in aqueous solution over a large concentration range. The extremely bright fluorescence of these nanoparticles, coupled with functionality for targeted cellular imaging, gives them enormous potential for fluorescence based imaging and sensing, possibly including applications with single nanoparticle detection limits.
Results and discussion
Preparation of PEG lipid-modified conjugated polymer nanoparticles
CPNs form in response to rapid dilution of conjugated polymer solutions into water; the hydrophobic polymer molecules collapse in aqueous solution to create nanoparticles with very high intrinsic fluorescence. To prepare CPNs which incorporate PEG lipid into the nanoparticle structure, conjugated polymer solutions in THF were diluted into aqueous solutions containing functionalized PEG lipid, during brief mild sonication to aid mixing, as described in detail in the Experimental section. The PEG lipid molecules used here contain two C14lipid chains linked to the PEG through a phosphate moiety and provide a bidentate hydrophobic group for interaction with conjugated polymer. The functional end group is located at the opposite end of the PEG chain.PEG lipid-CPNs were prepared using PFBT and a series of PEG lipids (PEG Mr = 2000, 1000, 550) with either carboxy, biotin, or methoxy end groups (Table 1). Our intent was to demonstrate that functionalized PEG lipid-CPNs can be prepared with a range of PEG sizes and moieties for bioconjugation, using a common strategy. Different end groups provide different moieties for conjugation to biomolecules; carboxy end groups allow for covalent linkage using established chemistry, and biotin end groups can be used to bind streptavidin or streptavidin-linked molecules with high affinity. We have also prepared PEG lipid-CPNs using other conjugated polymers, including PFO, PFPV, and MEH-PPV (Table S1, ESI†). These additional Nps behave similarly to PEG lipid-PFBT nanoparticles, with variations in size and spectral properties that most likely reflect the differences between their respective conjugated polymer starting materials (Table S1 and Fig. S2, ESI†).
The size of PFBT Nps formed in the presence of PEG lipid was characterized by both TEM and DLS. Representative TEM data and size distribution are shown in Fig. 1; TEM diameters obtained for PEG lipid-PFBT nanoparticles with different functional end groups are listed in Table 1. Additional TEM data and DLS data for PFBT Nps and those prepared with other conjugated polymers are included in the ESI† (Fig. S1 and Table S1). Mean PEG lipid-PFBT particle size is ca. 24 nm, and is insensitive to changes in the PEG lipid end groups and PEG Mr tested. DLS diameters for PEG lipid-PFBT Nps are 20–30 nm larger than those measured by TEM. Unlike TEM measurements, DLS size measurements reflect the hydrodynamic diameter and are expected to be somewhat higher than those measured by TEM, particularly for PEG coated nanoparticles, since extension of long PEG groups into solution will be accompanied by significant solvation not present under TEM conditions. Differences between TEM and DLS measurements observed here are similar to those reported for PEG coated semiconducting polymer nanospheres.22 However, absolute size values obtained by DLS measurements are accurate only for monodisperse particles; measured sizes can be inflated by the presence of even small amounts of aggregate. For this reason, we use DLS size measurements here as a tool for comparison of relative size and do not interpret DLS data in terms of absolute size.
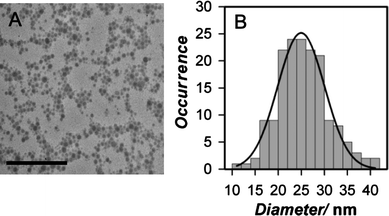 | ||
Fig. 1 Representative TEM images and size distribution for PEG lipid-PFBT nanoparticles. (A) TEM image of methoxy 2000 Mr PEG lipid-PFBT nanoparticles. Images were acquired at 120 kV on a cryostage at liquid nitrogen temperature and are shown at 120![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 magnification. Scale bar is 500 nm. (B) Histogram of size distribution and fit to a Gaussian function for the methoxy 2000 Mr PEG lipid-PFBT nanoparticles showing the average diameter to be 24 ± 5 nm (mean ± FWHM). 000 magnification. Scale bar is 500 nm. (B) Histogram of size distribution and fit to a Gaussian function for the methoxy 2000 Mr PEG lipid-PFBT nanoparticles showing the average diameter to be 24 ± 5 nm (mean ± FWHM). | ||
Using DLS, we also measured the ζ potential of the PEG lipid-PFBT Nps. These data provide initial evidence for PEG lipid incorporation into these CPNs. Since the ζ potential of PEG lipid CPNs reflects the charge of all constituent materials in Np, incorporation of different functionalized PEG lipid into the CPN will result in ζ potentials that reflect the charge of the different end groups, in addition to the phospholipid and constituent conjugated polymer. PFBT nanoparticles prepared with carboxy PEG lipid have negative ζ potential (−38 ± 1 mV), reflecting the negative charge on charged end group as well as the phospholipid, while biotin and methoxy PEG-lipid-CPNs have smaller negative ζ potentials (−9 ± 1 mV and −6 ± 1 mV, respectively) that reflect the neutral end group as well as the negative charge on the phospholipid.
Additional evidence for incorporation of PEG lipid into these CPNs comes from their observed properties, which are different than those of the corresponding unmodified CPNs. For example, PEG lipid-CPNs will pass through a size exclusion column in buffer (e.g. 30 cm G-25 Sephadex packed column, commonly used in separations for bioconjugation methods) with high recovery, while unmodified particles show strong nonspecific binding to the stationary phase. Similarly, CPNs prepared with PEG lipid can be filtered through hydrophobic membrane filters in buffer (e.g. 0.2 micron PVDF syringe filters) without difficulty; absorbance measurements of PEG lipid-CPNs before and after filtration are indistinguishable, consistent with no significant binding to the hydrophobic filter. In contrast, CPNs prepared without PEG lipid and diluted in buffer bind to the filter in small but visible quantities in our hands, either as a result of their higher hydrophobicity, possible instability in the presence of buffer salts, or the presence of aggregates. Most notably, we observe that PEG lipid-PFBT CPNs have higher quantum yield than the corresponding unmodified CPNs (Table 2). For example, methoxy 550 Mr PEG lipid-PFBT nanoparticles have a quantum yield of 19 ± 1%, compared to the value of 12 ± 1% we measure for unmodified particles prepared using the same conjugated polymer and conditions. On average, PEG-lipid-PFBT nanoparticles have nearly a 50% increase in quantum yield relative to unmodified nanoparticles. Together, these observations are consistent with incorporation of PEG lipid into the CPNs, with resulting increases in hydrophilicity and fluorescent brightness.
| Nanoparticle | Quantum yield |
|---|---|
| Methoxy 550 Mr PEG lipid-PFBT | 19 ± 1% |
| Biotin 2000 Mr PEG lipid-PFBT | 17 ± 1% |
| Carboxy 2000 Mr PEG lipid-PFBT | 18 ± 1% |
| PFBT (unmodified) | 12 ± 1% |
We hypothesize that PEG lipid-CPNs form via a process analogous to that proposed for unmodified CPNs formed viareprecipitation. When the conjugated polymer is diluted, nanoparticles experience a sudden change in the microenvironment of solvent, leading to collapse of the hydrophobic polymer chain into nanoparticles. We propose that in the presence of PEG lipid, the aliphatic side chains on the polymer backbone interact with the hydrophobic PEG lipid tail and are incorporated into the nanoparticle during chain collapse; the bidentate lipid tail inserts into the CPN core and is retained there by hydrophobic interactions, while the hydrophilic PEG group protrudes out into the aqueous solution. Hence, the CPN surface is modified with hydrophilic functionalized PEG that helps prevent aggregation, improves biocompatibility, and provides end groups that can be used for conjugation and labeling. A similar structure has been proposed for polymer-encapsulated quantum dot nanoparticles coated with PEG lipid.17,18
Our hypothesis of insertion of the lipid tail into the polymer chain during collapse is consistent with the observed higher quantum yield of the PEG lipid-CPNs relative to unmodified particles. It is known that conjugated polymer fluorescence is quenched by interactions between polymer fluorophores.23 For example, polymer aggregation lowers the quantum yields of conjugated polymer in aqueous or hydrophilic solutions. Similarly, unmodified CPNs have substantially lower quantum yields than their constituent conjugated polymer precursors, presumably due to interactions between polymer segments after chain collapse.24 As a result, the increase in quantum yield for PEG lipid-CPNs relative to unmodified CPNs can be rationalized on the basis of changes in the relative interactions of the polymer chain(s) in the CPNs caused by insertion of the lipid tail in the nanoparticle core; the lipid tails may create greater spacing between individual conjugated polymer fluorophores that contributes to reduced intrachain quenching and correspondingly larger quantum yields than those observed for unmodified CPNs. In this case, the absorbance maxima of PEG lipid CPNs should also be decreased relative to unmodified particles, since decreased interaction of polymer fluorophores is accompanied by blue shifts in the absorbance spectrum.25 Indeed, a comparison of high resolution absorbance spectra of methoxy 2000 Mr PEG lipid-CPNs and the corresponding particles prepared without PEG lipid demonstrates a decrease in absorbance maximum of 2.4 nm for PEG lipid CPNs (data not shown), consistent with disruption of interaction between conjugated polymer fluorophores by lipid insertion into the core.
We cannot rule out the possibility that PEG lipid-CPNs prepared here form by micelle entrapment of conjugated polymer nanoparticles, as proposed for PEG-capped polymer coated QDs20 and semiconducting polymer nanospheres.21 A study of micelle formation for 2000 MrPEG lipid reports a critical micellar concentration (CMC) value that is approximately micromolar, and a measured micelle size of ca. 17 nm.26 The PEG lipid concentrations used in our experiments (17–83 µM) are therefore above the CMC, and micelles may be present in solution prior to addition of conjugated polymer. However, the thermodynamic stability of PEG lipid micelles of such small size is predicted to be low,27,28 and our CPNs are not prepared under conditions that favor micelle formation. We carried out control experiments to investigate the presence of lipid micelles in our nanoparticle preparations; no measurable light scattering was observed in PEG lipid solutions at concentrations up to 83 µM (data not shown), indicating that micelle, if present, were not observable. Since the reported PEG lipid micelle size is comparable to or smaller than the reported diameter for unmodified CPNs (e.g. 10 to 30 nm for PFBT12), it is unlikely that partitioning of independently precipitated CPNs into preformed PEG lipid micelles could occur. If CPNs are present inside larger than predicted micelles, there must be intimate association of the lipid tails with the CPN structure sufficient to produce the observed increased quantum yield and blue shift in absorbance maximum. In this case, the final PEG lipid-CPN structure would be indistinguishable from that resulting from the proposed coprecipitation mechanism.
Optimization of PEG lipid-CPN preparation
To determine preparation conditions that result in maximal incorporation of CPNs with PEG lipid, experiments were carried out that altered the concentration of PEG lipid in solution. Initial nanoparticle preparations used 50 µg ml−1PEG lipid. However, PEG lipid-CPNs were also prepared using 25 µg ml−1, 100 µg ml−1, 150 µg ml−1, and 200 µg ml−1PEG lipid. No significant change in apparent hydrodynamic size of the resulting nanoparticles was observed by DLS relative to that for nanoparticles prepared in the original 50 µg ml−1PEG lipid concentration (Table 3). When nanoparticles were prepared in reduced concentrations of PEG lipid (less than 20 µg ml−1), a portion of the nanoparticle preparation bound to the membrane filter, reflecting increased hydrophobicity that presumably results from inadequate incorporation of PEG lipid into CPNs. These analyses suggest that maximum incorporation of the PEG lipids tested here is achieved for preparations that use a 50 µg ml−1PEG lipid solution, and increased PEG lipid concentrations have no effect.To see the dependence of PEG lipid-CPN size on initial polymer concentration, PEG lipid-PFBT nanoparticle solutions were prepared from initial conjugated polymer concentrations of 10–250 ppm in THFvia a ten-fold dilution to final concentrations ranging from 1 to 25 ppm (Table 4). The size of the resulting nanoparticles was evaluated by DLS. At these starting concentrations of conjugated polymer (10 to 250 ppm), the apparent hydrodynamic diameter of the PEG lipid-PFBT nanoparticles is independent of the starting concentration, and the only outcome of increased starting polymer concentration is increased nanoparticle concentration. However, at higher polymer concentrations, apparent particle size increases. Ten-fold dilutions of polymer concentrations above 500 ppm resulted in larger observed particle sizes by DLS. It has been observed that the size of unmodified CPNs also varies with starting polymer concentration,12 although no systematic study of this dependence has been reported.
Fluorescence properties of PEG lipid-CPNs
We evaluated the spectroscopic properties of PEG lipid-CPNs. Notably, the absorption and emission maxima of PEG lipid-CPNs (Fig. S2, ESI†) are close to those of particles prepared in the absence of PEG lipid for PFBT, PFO, PFPV, and MEH-PPV PEG lipid nanoparticles.12,24,29 Like unmodified CPNs the emission maxima of nanoparticles in aqueous solution are slightly red shifted compared to precursor conjugated polymer dissolved in organic solvent (THF), while the overall shape of the emission profile is maintained. This phenomenon has been attributed to a change in the spatial environment of the polymer fluorophores caused by folding to create the nanoparticle.24,29 As noted above, PEG lipid-CPNs are somewhat less red-shifted than bare particles (by 2.4 nm), presumably as a result of lipid insertion into the folded core. Like unmodified CPNs, PEG lipid-CPNs show good photostability (Fig. S3, ESI†). PEG lipid-PFBT nanoparticles show the best photostability in our experiments (Fig. S3†) which agrees with previous measurements of unmodified CPNs.12 Together, these data indicate that apart from increases in quantum yield, incorporation of PEG lipid does not substantially alter the spectroscopic behavior of CPNs.Stability of PEG lipid-CPNs in solution
Over time or at high concentrations, hydrophobic particles will tend to form small aggregates as a mechanism for water exclusion from hydrophobic surfaces, and minimization of aggregation is desirable to increase nanoparticle shelf life. Since incorporation of a PEG lipid into CPNs results in increased surface hydrophilicity, our expectation was that once formed, these PEG lipid-CPNs would be highly stable in solution. We have observed no signs of aggregation in any of the PEG lipid CPN solutions described here. However, to more thoroughly assess stability over time, the apparent diameter of CPNs in a 28 ppm solution of carboxy PEG lipid-PFBT nanoparticles was measured by DLS at intervals over 60 days (Fig. 2). No significant variation in apparent hydrodynamic size was observed, indicating that observable aggregation did not occur, and that these PEG lipid-CPNs are stable for long periods of time in solution. The resistance of these PEG lipid-CPNs to aggregation may reflect the surface charge contributed by the PEG lipid and end groups, steric effects of PEG interactions, and the increased hydrophilicity of the PEG. | ||
| Fig. 2 Apparent hydrodynamic size of biotin PEG lipid-PFBT nanoparticles as a function of time. Hydrodynamic size was monitored by DLS over 60 days. All measurements were in triplicate. Error bars are the standard deviation and are enclosed within the data symbol. Note that x-axis is not linear. | ||
Stability of PEG lipid-CPNs to concentration
Our standard protocol (10-fold dilution of 250 ppm conjugated polymer into 50 ppm PEG lipid) yields 28 ppm CPNs of reproducible size after evaporation of the THF. However, higher concentrations could be advantageous for specific applications, including chromatographic separations during bioconjugation and live cell experiments requiring dilution of CPN stock into media. As a result, we investigated the stability of PEG lipid nanoparticles to being concentrated by ultrafiltration with a centrifugal concentrator. In these experiments, a 28 ppm solution of carboxy PEG lipid-PFBT nanoparticle solutions was concentrated to a final concentration of 625 ppm. Portions of this concentrated solution were rediluted to 25 ppm before analysis by DLS. The resulting apparent hydrodynamic size (60 ± 2 nm) was indistinguishable from the size of original solutions (59 ± 2), indicating that no aggregation occurred as a consequence of concentration. No binding of CPN solutions to the concentrator filters could be observed. While we have not concentrated PEG lipid-CPNs above 625 ppm, we expect that even higher concentrations of these particles are achievable. In contrast, unmodified CPNs cannot be concentrated by ultrafiltration due to nonspecific binding to ultrafiltration membranes, and are currently concentrated by dilution in glycerol followed by vacuum evaporation30 to yield CPN solutions of nanoparticles in glycerol, with an upper concentration limit of about 200 ppm. Solutions concentrated via this method are not useful for cell studies, as glycerol is a potent osmolyte. The amenability of PEG lipid CPNs to concentration viaultrafiltration will allow their more widespread use in biological systems that require increased CPN concentration, or that require concentrations not achievable for unmodified CPNs.Bioconjugation of PEG lipid-CPNs
Conjugation of nanoparticles to specific biomolecules such as antibodies or other biomarkers is highly desired for specific labeling of biomolecules on or within the cell. The PEG lipid end groups incorporated into these CPNs contain inherent functionality for molecular recognition and/or covalent linkage. To demonstrate that these end groups are in a steric and conformational arrangement that allows bioconjugation, we carried out a series of experiments in PFBT PEG lipid-CPNs with biotin end groups were used to bind streptavidin. In the first set of experiments, biotin modified CPNs were incubated with magnetic streptavidin beads. After pulling out the beads from solution and washing to remove unbound nanoparticles, bound nanoparticles were removed by competition with free biotin and the magnetic beads removed with a strong permanent magnet. As shown in Fig. 3, the resulting supernatant contains significant nanoparticle fluorescence (solid line), indicative of biotin-functionalized PEG lipid-CPNs binding to the streptavidin beads. In contrast, no nanoparticle fluorescence was observed in the supernatant from magnetic beads incubated with carboxy-PEG lipid-CPNs (Fig. 3, dashed line), which lack the biotin functionality necessary for binding to the beads. These results demonstrate both successful incorporation of biotin PEG lipid into PFBT CPNs, and the availability of the PEG lipid end group for molecular recognition and/or covalent linkage.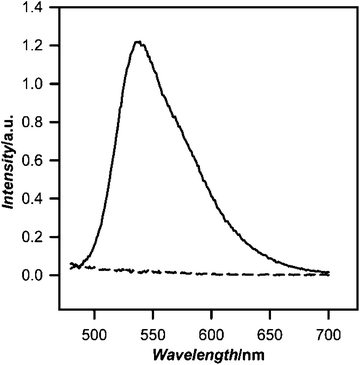 | ||
| Fig. 3 Fluorescence emission spectrum of biotinylated PEG lipid PFBT nanoparticle pull down with streptavidin magnetic beads. Streptavidin coated magnetic beads were incubated with biotinylated 2000 MrPEG lipid PFBT nanoparticles, washed to remove unbound CPNs, and then incubated with free biotin to release bound nanoparticles. Magnetic beads were removed with a strong magnet, and the fluorescence of the supernatant was recorded (solid line; λex = 460 nm). Control experiments using carboxy 2000 Mr PEG lipid-PFBT nanoparticles do not bind streptavidin magnetic beads, as shown by the emission spectrum of the control supernatant (dashed line). | ||
In additional experiments, a streptavidin coated cover glass was incubated with a very dilute solution of nanoparticles modified with biotin PEG lipid, rinsed to remove non-binding particles, and then air-dried. As shown in Fig. 4, significant numbers of near diffraction-limited spots of nanoparticle fluorescence can be observed (Fig. 4A), indicating biotin-functionalized PEG lipid CPNs binding to streptavidin on the glass surface. Spots with a range of brightness are observed, with brighter spots probably reflecting multiple nanoparticles bound to streptavidin clusters and/or nanoparticle clusters formed during drying. In contrast, very little nanoparticle fluorescence is observed in the control plates incubated with PEG lipid-CPNs with carboxy end groups (Fig. 4C). Together, these data indicate binding of biotin PEG lipid-CPNs to the streptavidin coated glass.
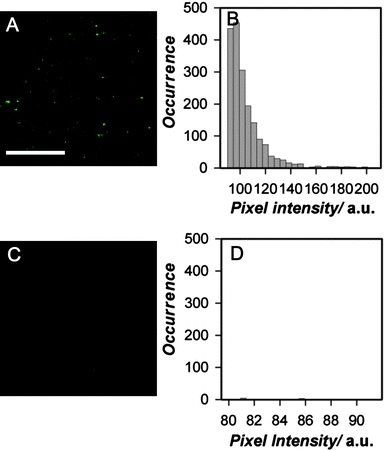 | ||
| Fig. 4 Single nanoparticle fluorescence and intensity distributions from biotinylated PEG lipid-PFBT nanoparticles bound to streptavidin coated cover glass. (A) Representative fluorescence image of streptavidin-coated cover glass incubated with biotinylated 2000 Mr PEG-lipid-PFBT nanoparticles (λex = 495 nm; λem = 510 nm long pass filter). Scale bar = 10 µm. (B) Histogram of biotinylated 2000 Mr PEG-lipid-PFBT nanoparticle fluorescence intensities obtained from the image; a threshold mask was applied to all objects. (C) Fluorescence image of streptavidin-coated glass slide incubated with carboxy 2000 Mr PEG lipid-PFBT nanoparticles, as a control for nonspecific binding to slide and (D) histogram of the 2000 Mr PEG lipid-PFBT nanoparticle fluorescence intensities obtained from the image. Exposure times were identical for (A) and (C). The diffraction limit of the microscope was 225 nm. | ||
Surprisingly, careful examination of the CPN signal observed in these cover glass experiments suggests possible observation of single nanoparticles. The diffraction limit of our microscope is 225 nm. Hence, we cannot distinguish the signal from individual nanoparticles if they are a distance of less than 225 nm apart. However, variations in the intensity of individual sites of PFBT fluorescence can be used to indicate varying numbers of CPNs in individual diffraction-limited spots. We estimate that a signal from a single nanoparticle of diameter 25–50 nm (TEMvs.DLS diameter) could occupy one to four pixels in these images, depending on whether the nanoparticle was located in the center or periphery of individual pixels. We examined the intensity of all image objects occupying four or fewer pixels. As shown in Fig. 4B, the intensity distribution of the near diffraction-limited regions shows a narrow distribution of biotin PEG lipid-CPN spots of approximately constant intensity (Fig. 4B), consistent with measurement of single particles. We cannot distinguish between single particles and small aggregates of consistent size under these conditions. However, given the lack of aggregation evident in the TEM data for these 2000 Mr PEG lipid-PFBT Nps (Fig. 1), the observed size stability of these nanoparticles in solution over time (Fig. 2) and the precedent for nonaggregation of PEG coated particles in previously published systems,19,31 formation of aggregates is not expected here. A substantive conclusion of single particle imaging under these conditions requires additional experimentation. However, the possibility of single particle imaging data obtained here with a standard camera and arc lamp excitation highlights the extreme brightness of these PEG lipid-CPNs and their potential utility for single particle imaging in biological systems.
Targeting of bioconjugated PEG lipid CPN to CD16/32 receptors on the cell surface
The properties of PEG lipid-CPNs such as their high extinction coefficient, bright fluorescence, photostability, and functionalization indicate significant potential for targeted single particle imaging and tracking in living cells. Here we demonstrate targeted localization of functionalized nanoparticles to individual CD16/32 receptors on the surface of mouse macrophageJ774A.1 cells. In these experiments, a commercially available biotin-linked rat anti-CD16/32 antibody was bound to CD16/32 on the cell surface, and then labeled with biotin-functionalized PEG lipid PFBT nanoparticles, using streptavidin as a linker in a sandwich format. The result was Ab-conjugated nanoparticles that specifically labeled antibody-tagged receptors on the cell surface. Fig. 5 shows the differential interference contrast and fluorescence images taken of labeled cells. Localized nanoparticle fluorescence is observed on the periphery of the cell, as is typical for membrane localization. Control experiments performed either without streptavidin or using carboxy modified nanoparticles instead of biotinylated nanoparticles showed no fluorescence (data not shown), indicating that observed binding of biotinylated PEG lipid-PFBT Nps does not represent nonspecific adsorption to the cell membrane. Together, these data demonstrate that PEG lipid-CPNs can target specific tagged proteins on the cell surface. To our knowledge, these results represent the first report of targeted delivery of CPNs to individual sites on the cell surface. | ||
| Fig. 5 Biotinylated PEG lipid-PFBT nanoparticles targeted to cell surface receptors. (A) Differential Interference Image (DIC) of fixed J774A.1 cells; (B) fluorescence image of fixed J774A.1 cells labeled with biotinylated PFBT nanoparticles. Scale bar is 25 µm. J774 A1 macrophage cells which express CD16/32 (Fc receptor) were paraformaldehyde fixed and incubated with biotinylated anti-CD16/32 antibody. After washing the cell with RB, the cells were next incubated with streptavidin, washed, and labeled with biotinylated PEG lipid-PFBT nanoparticles. Images were obtained with 495 nm excitation, using a 510 nm long pass emission filter. | ||
Experimental
Reagents
The polyfluorene conjugated polymers PFBT (poly[(9,9-dioctylfluorenyl-2, 7-diyl)-co-(1,4-benzo-{2,1′,3}-thiadiazole)], Mw 48000, polydispersity 2.7), PFPV (poly[(9,9-dioctyl-2,7-divinylenefluorenylene)-alt-co-{2-methoxy-5-(2-ehtylhexyloxy)-1,4-phenylene}], Mw 85000, polydispersity 5.4), MEH-PPV (poly[2-methoxy-5-(2-ethylhexyloxy)-1,4-phenylenevinylene]-end capped with DMP, Mw 565000, polydispersity 5.1) and PFO (poly[(9,9-dioctylfluorenyl-2,7-diyl)]-end capped with DMP, Mw 29000, polydispersity 3.0) were purchased from American Dye source, Inc (Quebec, Canada). Mr 2000 PEG lipids with biotin end groups (1,2-dimyristoyl-sn-glycerol-3-phosphoethanolamine-N-[biotinyl (polyethylene glycol)-2000]); (Ammonium salt)), and carboxy end groups (1,2-dimyristoyl-sn-glycerol-3-phosphoethanolamine-N-[carboxy (polyethylene glycol)-2000]); (ammonium salt) and 550, 1000, and 2000 MrPEG lipid with methoxy end groups (1,2-dimyristoyl-sn-glycerol-3-phosphoethanolamine-N-[methoxy (polyethylene glycol)-550, 1000, or 2000]) were purchased from Avanti Polar Lipids. THF (anhydrous HPLC grade) was obtained from Fisher Scientific. Biotin rat anti-mouse CD16/CD 32 antibody was purchased from BD Pharmingen. All chemicals and biological molecules were used without further purification.
Method for preparation of nanoparticles
PFO, PFPV, PFBT, and MEH-PPV CPNs were prepared with biotin, carboxy, amine, and methoxy functionalized PEG lipids (550, 1000, or 2000 Mr PEG). Stock solutions of conjugated polymers (1000 ppm) were dissolved in HPLC grade THF by stirring overnight. Next, functionalized PEG lipids were dissolved in distilled-deionized H2O (ddH2O) at concentrations between 25 and 250 ppm. The conjugated polymer nanoparticles were then prepared by rapidly dispersing 1 ml of conjugated polymer solution (10–250 ppm) into 9 ml of the PEG lipid solution under continuous mild sonication (45% amplitude) with a microtip-equipped probe sonicator (Branson, 4C15) for two minutes. THF was evaporated under vacuum overnight at room temperature. Finally, the nanoparticles were filtered through a 0.2 µm polyvinylidene fluoride (PVDF) syringe filter (National Scientific).Nanoparticle characterization
The nanoparticles were evaluated by transmission electron microscopy (TEM) and dynamic light scattering. A Hitachi H7600T TEM at 120 kV and a cryostage at liquid nitrogen temperatures were used for all TEM measurements. Samples were prepared by drop casting nanoparticle solutions onto Formvar/carbon grids. CPN diameter was measured with Image J. The measured particle diameters were fit to a Gaussian distribution using SigmaPlot (Systat).Dynamic light scattering was performed using a Malvern Zetasizer (ZS90) at 25 °C using distilled-deionized H2O (ddH2O) as dispersant. Prior to each DLS measurement, samples were briefly sonicated in a bath sonicator for 30 seconds to remove bubbles and minimize aggregates. The Z-average and polydispersity index were determined using cumulants analysis and manufacturer supplied software. Data were analyzed in terms of intensity weighted distributions. Three runs were performed for each sample, and the mean and standard deviation of both the Z-average and polydispersity index were calculated.
Fluorescence emission spectra of the CPNs were acquired using a photon counting spectrofluorometer (Photon Technology International; QM-4). The fluorescence emission of PFBT nanoparticles was measured from 480 nm to 700 nm in aqueous solution using 460 nm excitation. PFO nanoparticles were excited at 384 nm and emission measured from 395 to 700 nm. PFPV nanoparticles were excited at 458 nm and emission measured from 480 to 700 nm. MEH-PPV nanoparticles were excited at 498 nm and emission measured from 510 to 725 nm. Both excitation and emission monochromator slits were set to achieve a 4 nm band pass. Absorbance spectra were recorded using a Genesys 10UV Scanning spectrophotometer (Thermo) using a 1 cm quartz cuvette. Individual quantum yields were calculated using fluorescein in 0.1 M NaOH as a standard. High resolution spectra of methoxy 2000 Mr PEG lipid-PFBT and bare PFBT particles were acquired using a UV-2501PC (Shimadzu) scanning spectrophotometer with 0.5 nm spectral resolution.
Nanoparticle concentrations were estimated from the mass of conjugated polymer starting material diluted into aqueous solution, assuming complete polymer to nanoparticle conversion. Specifically, nanoparticle volumes were calculated from the particle diameter measured by TEM, assuming a spherical shape and converted to nanoparticle mass assuming a nanoparticle density of 1 g cm−3 (actual density is between 0.95 and 1.05 g cm−3); dividing the total mass of conjugated polymer used in the reprecipitation by the mass of a single nanoparticle then yielded the number of nanoparticles formed, which was easily converted to moles of nanoparticles and molar concentration of the nanoparticle suspension. Concentration calculations do not take into account small reductions in yield that result from filtration and may therefore be a slight overestimate. UV measurements taken before and after filtration indicate that any loss from filtration, if any, is small, and demonstrate negligible formation of aggregates.
Concentrating PEG lipid modified nanoparticles
Solutions containing dilute PEG lipid-CPNs were concentrated by ultrafiltration using a 30 kDa cutoff centrifugal concentrator with a regenerated cellulose filter (Millipore) according to manufacturer protocols. Solutions were concentrated up to 625 ppm.Streptavidin pull-down of biotin PEG lipid-PFBT nanoparticles
PFBT nanoparticles (28 ppm) prepared with biotin functionalized PEG lipid were incubated with magnetic streptavidin beads (New England Biolabs) for 30 min in phosphate buffered saline (PBS; 140 mMNaCl, 2.7 mMKCl, 10 mMNa2HPO4, 1.8 mMKH2PO4). The magnetic beads were removed from solution using a strong permanent magnet and washed 5× in PBS to remove any unbound CPNs. The magnetic beads were then incubated overnight with 0.2 mg ml−1 free biotin to dissociate the CPNs from the magnetic beads. The magnetic beads were then removed from solution using a strong permanent magnet. The fluorescence from the remaining biotinylated nanoparticles was measured using 460 nm excitation while scanning the fluorescence emission from 480 to 700 nm. The slits for both the excitation and emission monochromators were set to achieve a 2 nm band pass.Imaging of biotinylated nanoparticles localized on streptavidin-coated cover glass
For single particle fluorescence imaging, cover glasses were cleaned with concentrated sulfuric acid, washed with water and dried in air. Clean cover glasses were then coated with 1% poly-L-lysine, washed with water to remove excess poly-L-lysine, incubated with 1 mg ml−1 streptavidin for 30 minutes, and carefully washed with Ringer's buffer 3× (RB; 155 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 2 mM NaH2PO4, 10 mM glucose, 10 mM HEPES, pH 7.2). Dilute solutions (5 pM/25 ppb) of CPNs (carboxy PEG lipid-PFBT and biotin PEG lipid-PFBT) were incubated with the streptavidin-modified cover glass by inverting the cover glass over a drop of dilute CPNs on parafilm. Cover glasses were incubated with PEG lipid-CPNs for half an hour, washed carefully 3× with RB buffer, and air dried before taking images. Fluorescence imaging was performed by inverted epifluorescence microscope (Olympus IX71, 60×/1.45 N.A. objective using Xe arc lamp excitation, 495/10 nm excitation filter, and 510 nm long pass emission filter).Targeting of PEG lipid-PFBT nanoparticles to cell surface receptors
J774A.1 macrophage cells were plated onto 35 mm glass bottom microscope dishes in DMEM (Dulbecco's Modified Eagle's Medium) containing 10% fetal bovine serum, 1% penicillin–streptomycin and 1% glutamate and incubated in humidified environment overnight (5% CO2, 37 °C). Adherent cells were washed with RB 3×, fixed with 4% paraformaldehyde for 10 minutes at 37 °C, and blocked with 1% bovine serum albumin (BSA) for 1 hour at room temperature, washed again, and treated with 1:1000 dilution of biotin rat anti-mouse CD16/CD32 antibodies (BD Pharmingen) for 2 hours. Cells were washed 3× with RB to remove unbound antibodies before incubating with 1 µg ml−1 streptavidin for 30 minutes, again at room temperature. After streptavidin incubation, cells were washed 3× with RB, then incubated with PEG lipid-CPNs (biotin 2000 Mr PEG lipid-PFBT or carboxy 2000 Mr PEG lipid-PFBT; 5 pM/25 ppb) for 30 minutes, followed by an additional three washes with RB. Images were acquired with an inverted microscope [Olympus IX71, Xe arc lamp for excitation and filters and beam splitters (495/10 for excitation and 510 LP for emission) from Chroma, Ocra-ER CCD (Hamamatsu)]. The acquired images were analyzed using Slidebook 5.0.
Conclusions
Incorporating functionalized PEG lipids into CPNs is a simple method for preparing extremely bright biocompatible nanoparticles with enhanced properties suitable for fluorescence imaging applications. The PEG lipids impart improved hydrophilicity and quantum yield, and straightforward conjugation to biomolecules for targeted delivery. We have demonstrated the utility of bioconjugation via PEG lipid biotin end groups. The resulting data demonstrate that functional end groups on PEG lipid-CPNs provide a platform to conjugate nanoparticles to molecules of biological importance. Hence, PEG lipid-CPNs are a viable technology for a wide range of labeling and imaging applications in living biological systems.Acknowledgements
The authors thank Dr George Chumanov for use of his high resolution spectrophotometer and Dr Jason McNeill for helpful discussion of the manuscript. Financial support for this project came from the National Institutes of Health (1R01GM081040).References
- A. Alivisatos, W. Gu and C. Larabell, Annu. Rev. Biomed. Eng., 2005, 7, 55–76 CrossRef CAS.
- W. Hild, M. Breunig and A. Goepferich, Eur. J. Pharm. Biopharm., 2008, 68, 153–168 CrossRef CAS.
- J. Yan, M. Estevez, J. Smith, K. Wang, X. He, L. Wang and W. Tan, Nano Today, 2007, 2, 44–50 CrossRef.
- J. West and N. Halas, Annu. Rev. Biomed. Eng., 2003, 5, 285–292 CrossRef CAS.
- K. Thurn, E. Brown, A. Wu, S. Vogt, B. Lai, J. Maser, T. Paunesku and G. Woloschak, Nanoscale Res. Lett., 2007, 2, 430–441 CrossRef CAS.
- J. Gao and B. Xu, Nano Today, 2009, 4, 37–51 CrossRef.
- C. Kirchner, T. Liedl, S. Kudera, T. Pellegrino, A. Javier, H. Gaub, S. Stolzle, N. Fertig and W. Parak, Nano Lett., 2005, 5, 331–338 CrossRef CAS.
- A. Derfus, W. Chan and S. Bhatia, Nano Lett., 2004, 4, 11–18 CrossRef CAS.
- N. Lewinski, V. Colvin and R. Drezek, Small, 2008, 4, 26–49 CrossRef CAS.
- A. Hoshino, K. Fujioka, T. Oku, M. Suga, Y. Sasaki, T. Ohta, M. Yasuhara, K. Suzuki and K. Yamamoto, Nano Lett., 2004, 2163–2169 CrossRef CAS.
- C. F. Wu, C. Szymanski and J. McNeill, Langmuir, 2006, 22, 2956–2960 CrossRef CAS.
- C. Wu, B. Bull, C. Szymanski, K. Christensen and J. McNeill, ACS Nano, 2008, 2, 2415–2423 CrossRef CAS.
- L. Fernando, P. Kandel, J. Yu, J. McNeill, P. Ackroyd and K. Christensen, Biomacromolecules, 2010, 11, 2675–2682 CrossRef CAS.
- P. Howes, R. Thorogate, M. Green, S. Jickells and B. Daniel, Chem. Commun., 2009, 2490–2492 RSC.
- K. Li, J. Pan, S. Feng, A. Wu, K. Pu, Y. Liu and B. Liu, Adv. Funct. Mater., 2009, 19, 3535–3542 CrossRef CAS.
- J. Dubach, D. Harjes and H. Clark, J. Am. Chem. Soc., 2007, 129, 8418–8419 CrossRef CAS.
- A. Smith, H. Duan, M. Rhyner, G. Ruan and S. Nie, Phys. Chem. Chem. Phys., 2006, 8, 3895–3903 RSC.
- W. Yu, E. Chang, J. Falkner, J. Zhang, A. Al-Somali, C. Sayes, J. Johns, R. Drezek and V. Colvin, J. Am. Chem. Soc., 2007, 129, 2871–2879 CrossRef CAS.
- E. Chang, N. Thekkek, W. Yu, V. Colvin and R. Drezek, Small, 2006, 2, 1412–1417 CrossRef CAS.
- B. Dubertret, P. Skourides, D. Norris, V. Noireaux, A. Brivanlou and A. Libchaber, Science, 2002, 1759–1762 CrossRef CAS.
- P. Howes and M. Green, Photochem. Photobiol. Sci., 2010, 9, 1159–1166 RSC.
- P. Howes, M. Green, J. Levitt, K. Suhling and M. Hughes, J. Am. Chem. Soc., 2010, 132, 3989–3996 CrossRef CAS.
- G. Padmanaban and S. Ramakrishnan, J. Phys. Chem. B, 2004, 108, 14933–14941 CrossRef CAS.
- C. Wu, C. Szymanski and J. McNeill, Langmuir, 2006, 22, 2956–2960 CrossRef CAS.
- B. Schwartz, Annu. Rev. Phys. Chem., 2003, 54, 141–172 CrossRef CAS.
- B. Ashok, L. Arleth, R. Hjelm, I. Rubinstein and H. Onyüksel, J. Pharm. Sci., 2004, 93, 2476–2487 CrossRef CAS.
- V. Torchilin and V. Weissig, Liposomes, Oxford University Press, New York, 2 edn, 2003 Search PubMed.
- M. Johnsson, P. Hansson and K. Edwards, J. Phys. Chem. B, 2001, 105, 8420–8430 CrossRef.
- C. Szymanski, C. Wu, J. Hooper, M. Salazar, A. Perdomo, A. Dukes and J. McNeill, J. Phys. Chem. B, 2005, 109, 8543–8546 CrossRef CAS.
- J. Yu, C. Wu, S. Sahu, L. Fernando, C. Szymanski and J. McNeill, J. Am. Chem. Soc., 2009, 131, 18410–18414 CrossRef CAS.
- B. Dubertret, P. Skourides, D. Norris, V. Noireaux, A. Brivanlou and A. Libchaber, Science, 2002, 298, 1759–1762 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional TEM data, supplemental light scattering measurements, absorbance and fluorescence emission spectra, and photostability measurements. See DOI: 10.1039/c0nr00746c |
| This journal is © The Royal Society of Chemistry 2011 |